El síndrome de Von Hippel-Lindau se debe a mutaciones en el gen VHL y se caracteriza por hemangioblastomas del cerebro, la médula espinal y la retina del ojo. Además, los individuos afectados tienen un mayor riesgo de carcinoma de células renales y quistes renales, feocromocitoma, quistes y tumores neuroendocrinos del páncreas, tumores del saco endolinfático y quistes del epidídimo, así como de los ovarios y las trompas de Falopio.
Los síntomas dependen del tamaño y la localización de los tumores [1]. Los niños pueden sufrir dolores de cabeza y sentirse mareados o débiles. Además, pueden producirse alteraciones visuales, que pueden llevar a la pérdida de visión en tumores crecientes de la retina, e hipertensión. Puede haber una pérdida de coordinación. Alrededor del 10% de los niños afectados tienen un tumor en el oído interno, que puede afectar a la audición. Sin tratamiento, los enfermos pueden quedarse ciegos, sufrir daños cerebrales o morir. Las muertes suelen ser el resultado de complicaciones de angiomas cerebrales o cáncer de riñón.

El diagnóstico precoz es de gran importancia. Fenotípicamente, se distinguen diferentes tipos de síndrome de Von Hippel-Lindau [1]: El tipo I se caracteriza por la aparición de hemangiomas en la retina y/o el sistema nervioso central, carcinomas de células renales y/o tumores neuroendocrinos. Sin embargo, el riesgo de feocromocitoma es muy bajo. El tipo I se asocia a mutaciones sin sentido o a deleciones de segmentos génicos más grandes. Por el contrario, el tipo II se asocia a menudo con mutaciones missense y el riesgo de desarrollar feocromocitomas es muy elevado.
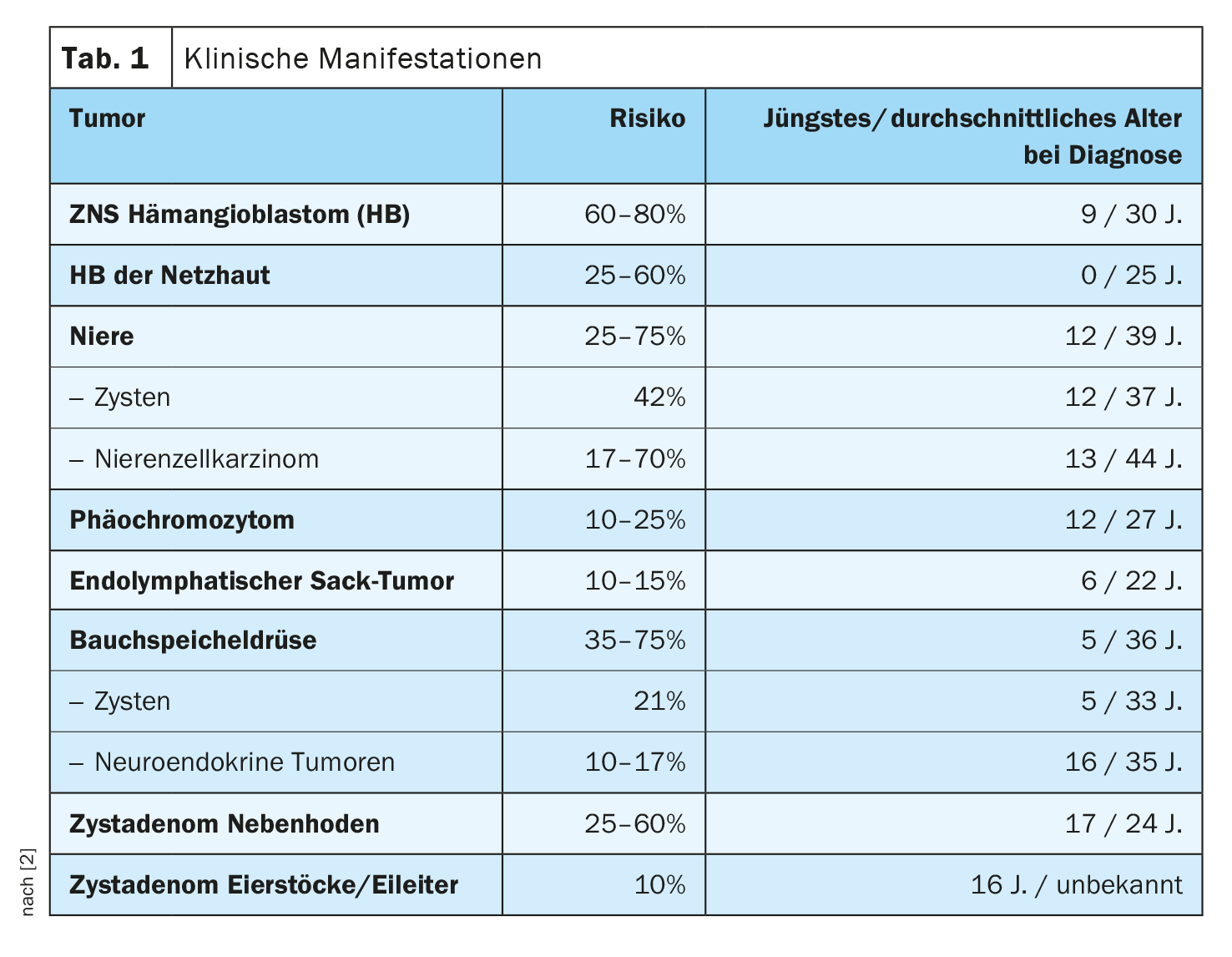
Criterios diagnósticos
El diagnóstico del síndrome de Von Hippel-Lindau se considera confirmado si se detecta una mutación en el gen VHL y/o [2]:
Sin síndrome de Von Hippel-Lindau en la familia en presencia de al menos 2 de las siguientes conclusiones:
≥2 hemangioblastomas de la retina, la médula espinal o el cerebro, o un único hemangioblastoma junto con una manifestación en el abdomen (por ejemplo, quistes múltiples de los riñones o el páncreas).
– Carcinoma de células renales
– Feocromocitoma
– ELST, cistadenoma de los ovarios o de las trompas de Falopio/epidídimo o tumores neuroendocrinos del páncreas
Con síndrome de Von Hippel-Lindau en la familia en presencia de al menos. 1 de las siguientes conclusiones:
– Hemangioblastoma de retina
– Hemangioblastoma de la médula espinal o del cerebelo
– Feocromocitoma
– Carcinoma de células renales
– Múltiples quistes en los riñones o el páncreas
Literatura:
- Institut für Klinische Genetik, Universitätsklinikum Carl Gustav Carus Dresden, www.uniklinikum-dresden.de, (última consulta 17.01.2023)
- Medizinische Hochschule Hannover, www.krebs-praedisposition.de/fuer-patienten-und-familien/von-hippel-lindau-syndrom/#diagnose, (última consulta 17.01.2023)
- Baumgartner-Parzer S: J Klin Endokrinol Stoffw 2020; 13: 37–40.
HAUSARZT PRAXIS 2023; 18(1): 47
InFo ONKOLOGIE & HÄMATOLOGIE 2023; 11(1): 32