
La pérdida de función del canal regulador de la conductancia transmembrana de la fibrosis quística (CFTR) conduce a la formación de una secreción muy viscosa en la fibrosis quística, lo que provoca una reducción del aclaramiento mucociliar. Los pacientes refieren tos crónica y esputo purulento, así como un aumento de las infecciones respiratorias. Pueden producirse atelectasias debido a la obstrucción de las secreciones; otras complicaciones pueden ser neumotórax o hemoptisis.
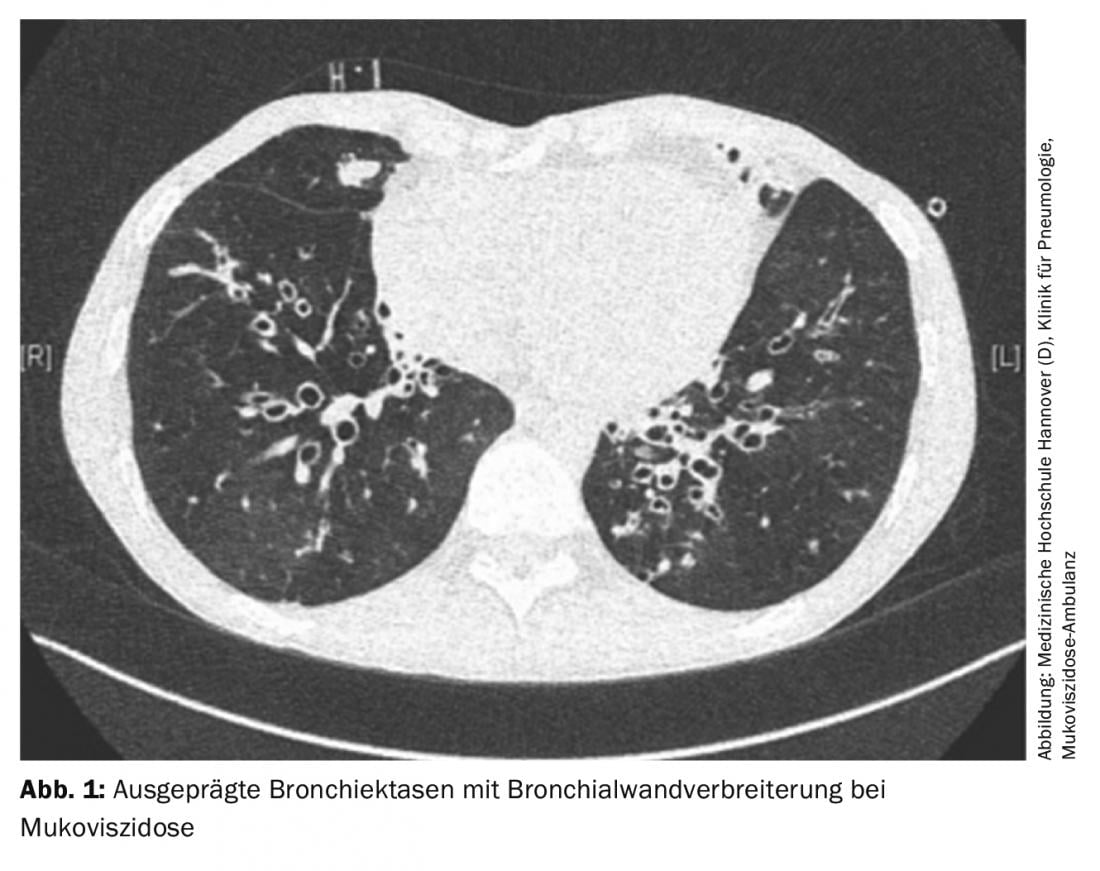
La pérdida de función del canal regulador de la conductancia transmembrana de la fibrosis quística (CFTR) conduce a la formación de una secreción muy viscosa en la fibrosis qu ística, lo que provoca una reducción del aclaramiento mucociliar. Esto favorece la colonización bacteriana, lo que conduce a la inflamación y destrucción del tejido pulmonar con la consiguiente insuficiencia respiratoria. Desde el punto de vista funcional pulmonar, a medida que avanza la enfermedad, suele haber un trastorno obstructivo de la ventilación con hiperinsuflación. Los pacientes refieren tos crónica y esputo purulento, así como un aumento de las infecciones respiratorias. El diagnóstico por imagen suele mostrar bronquiectasias llenas de moco con paredes bronquiales engrosadas (Fig. 1). Pueden producirse atelectasias debido a la obstrucción de las secreciones; otras complicaciones pueden ser neumotórax o hemoptisis.
En función de estos síntomas, el tratamiento de la enfermedad pulmonar se centra en la secretolisis y la terapia antibacteriana y antiinflamatoria.
Secretolisis
Moduladores del CFTR: El canal CFTR es un canal aniónico para el cloruro y el bicarbonato. Esta proteína unida a la membrana se localiza en el epitelio respiratorio, pero también en el tracto gastrointestinal y en muchos otros órganos. El primer modulador del CFTR, el ivacaftor, fue aprobado hace 10 años como la primera terapia que afecta al defecto de base subyacente de la fibrosis quística (Tabla 1).
Ivacaftor
Los moduladores CFTR se distinguen entre potenciadores y correctores. Los potenciadores, entre los que se encuentra el ivacaftor, aumentan la probabilidad de apertura del canal CFTR mediante la activación de mecanismos independientes del ATP. En consecuencia, los estudios clínicos mostraron un efecto en las mutaciones en las que el canal iónico CFTR está presente en la membrana celular pero es necesario aumentar la probabilidad de apertura. El FEV1 aumentó 10,6% puntos en los pacientes con al menos una mutación G551D (c.1652G>A). La probabilidad de sufrir una exacerbación pulmonar disminuyó en un 55%, y la calidad de vida y el peso aumentaron.
Estos resultados condujeron a la aprobación del ivacaftor en 2012 para las mutaciones gating, que sin embargo son raras, por lo que alrededor del 7% de los pacientes con FQ podrían recibir el fármaco. Los estudios de registro de Gran Bretaña y EE.UU. muestran que los efectos positivos siguen siendo detectables 4 y 5 años después del inicio de la terapia, respectivamente.
Lumacaftor
En aproximadamente el 50% de los pacientes con FQ, la mutación F508del es homocigota. En este grupo tan numeroso, la terapia con ivacaftor no había resultado eficaz. Los estudios de fase 2 indicaron que una combinación de ivacaftor con lumacaftor podría producir una mejora clínica. El lumacaftor pertenece a los correctores del CFTR. Aumenta la cantidad de proteína CFTR en la membrana celular cambiando el procesamiento en la célula.
En los estudios de fase 3, esta terapia combinada mostró un efecto significativo, aunque menor que el efecto del ivacaftor sobre las mutaciones gating. El FEV1 aumentó un 3% de puntos y las exacerbaciones disminuyeron un 30%. Esta terapia fue aprobada para este grupo de pacientes en 2016.
Tezacaftor
Dos años más tarde, se publicaron datos sobre otro modulador del CFTR, el corrector tezacaftor en combinación con ivacaftor. En un estudio de fase 3 controlado con placebo en pacientes con mutación homocigota F508del, el tratamiento combinado de tezacaftor e ivacaftor mostró superioridad con un aumento del 4% en puntos del FEV1. De nuevo, la tasa de exacerbaciones pulmonares fue un 35% menor que en el grupo placebo. Así pues, el efecto fue comparable al del Orkambi en pacientes homocigotos F508del.
Otro estudio comparó la eficacia en pacientes que tenían F508del y otra mutación CFTR con función residual. Estos pacientes se dividieron en tres grupos: un grupo recibió tezacaftor en combinación con ivacaftor, el segundo grupo recibió sólo ivacaftor y el tercer grupo recibió un placebo. Ambos grupos de tratamiento fueron superiores al grupo placebo en términos de FEV1. El Ivacaftor por sí solo produjo un aumento de 4,7% puntos, el tratamiento combinado alcanzó incluso un aumento de 6,8% puntos.
Basándose en estos resultados, el tratamiento combinado se aprobó tanto para los pacientes homocigotos F508del como para los pacientes que tienen F508del y otra mutación CFTR con función residual.
Elexacaftor/Tezacaftor/Ivacaftor
Otro gran paso en la mejora de la terapia queda patente en los ensayos clínicos sobre la eficacia de la triple combinación de elexacaftor, tezacaftor e ivacaftor. En un estudio controlado con placebo publicado en 2019, se estudió a pacientes que tenían un F508del y otra mutación CFTR con función mínima (aproximadamente 200 mutaciones diferentes).
Los resultados de este estudio superaron claramente los hallazgos anteriores. La función pulmonar mostró un aumento del FEV1 tras sólo 2 semanas de tratamiento, que se mantuvo estable en 13,6% puntos hasta la semana 24, y la tasa de exacerbaciones pulmonares se redujo en un 60%. La calidad de vida de los pacientes aumentó significativamente (18,1 puntos en el CFQ-R), al igual que su peso (IMC +1,3). Para evaluar la función CFTR durante la terapia, se realizaron pruebas de sudor. Se produjo un descenso significativo de 40 mmol/l en la concentración de cloruro en sudor, que está patológicamente aumentada en la fibrosis quística, de modo que muchos pacientes ya no superaban el límite de 60 mmol/l, necesario para el diagnóstico de la fibrosis quística.
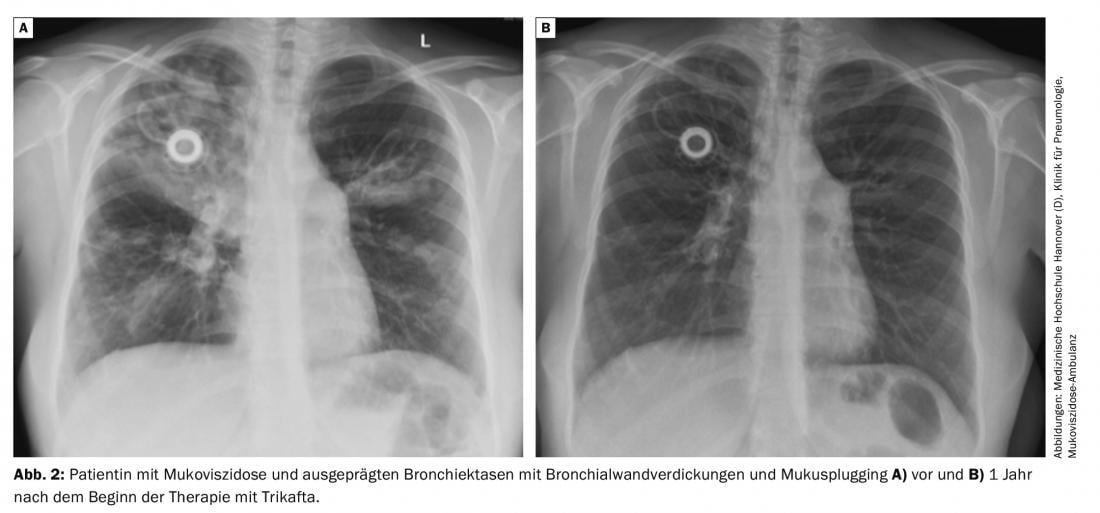
La eficacia de la triple combinación se probó adicionalmente en pacientes homocigotos F508del. La terapia con tezacaftor e ivacaftor sirvió de control en este estudio, que sólo duró 4 semanas. La combinación triple fue claramente superior a la terapia con tezacaftor e ivacaftor, el FEV1 aumentó un 10% de puntos, y también hubo claras ventajas en la calidad de vida y en el contenido de cloruro de la prueba del sudor (fig. 2) .
La terapia con elexacaftor/tezacaftor/ivacaftor suele tolerarse bien. Los acontecimientos adversos que se produjeron con mayor frecuencia en el grupo de verum que en el de placebo durante el ensayo clínico fueron erupciones cutáneas (10%), aumento de las enzimas hepáticas (10%), dolor de cabeza y diarrea. El 1% de los pacientes interrumpieron el tratamiento debido a acontecimientos adversos.
Los moduladores CFTR deben tomarse con una comida que contenga grasa. Los pacientes no deben comer pomelo o naranjas amargas ni tomar hierba de San Juan debido a las interacciones, y la dosis debe ajustarse en caso de uso simultáneo de inhibidores moderados o fuertes del CYP3A (por ejemplo, antifúngicos azólicos, antidepresivos).
El coste de la terapia con moduladores de CFTR es de 12.000-17.000 francos suizos al mes. Por lo tanto, la indicación, el inicio y el seguimiento de la terapia deben correr a cargo de un centro con experiencia en el tratamiento de la fibrosis quística.
Detrás de estas impresionantes cifras se esconde un cambio fundamental en la calidad de vida y la perspectiva vital en la vida cotidiana. Muchos pacientes ya no tienen tos ni expectoración, síntomas que antes eran compañeros constantes. Aunque no existen datos sobre la evolución a largo plazo de la enfermedad, cabe suponer que la esperanza de vida aumenta significativamente con estos fármacos.
Sin embargo, la medicación no funciona igual en todos los pacientes y los pacientes con pulmones gravemente destruidos siguen teniendo una función pulmonar deteriorada. La terapia no está aprobada para aproximadamente el 10% de los pacientes y aproximadamente el 1-2% de los pacientes interrumpen la terapia debido a intolerancias. Por lo tanto, otras formas de terapia siguen siendo importantes.
Dornasa alfa: En el contexto de la inflamación de las vías respiratorias, el ADN se libera a través de la desintegración de los granulocitos. La dornasa alfa escinde el ADN y reduce así la viscosidad de la secreción. En los ensayos clínicos, esto se tradujo en una reducción de las exacerbaciones pulmonares y una mejora de la función pulmonar.
Solución salina hipertónica y manitol: Un efecto osmótico provoca una reducción de la viscosidad del esputo tras la inhalación de solución salina hipertónica o manitol, lo que mejora el aclaramiento mucociliar. Para evitar la obstrucción de las vías respiratorias, debe utilizarse un broncodilatador antes de la inhalación.
Fisioterapia y deporte: La fisioterapia y el deporte forman parte de la terapia básica, aunque no existen suficientes estudios clínicos que demuestren su eficacia. La terapia consiste en aprender técnicas para mejorar el aclaramiento mucociliar, optimizar la tos y la técnica de inhalación, la movilización del tórax y el entrenamiento de fuerza y resistencia.
Terapias antibióticas
Staphylococcus aureus: En niños y adolescentes, el Staphylococcus aureus es el germen que se detecta con más frecuencia en las secreciones bronquiales. No existe una estrategia uniforme para tratar este germen. Hay indicios de que se produce una mayor inflamación de las vías respiratorias por este germen y que, por tanto, tiene importancia para el curso de la enfermedad. Por ello, a menudo se lleva a cabo un intento de erradicación con antibióticos orales, especialmente en niños, incluso en caso de detección asintomática. En caso de detección sintomática, debe administrarse en cualquier caso una terapia antibiótica oral durante 2-3 semanas.
Con el aumento de la edad, suelen observarse indicios crónicos de Staphylococcus aureus. Sin embargo, la terapia estafilocócica a largo plazo sólo debe considerarse en casos excepcionales, por ejemplo, exacerbaciones pulmonares frecuentes, ya que hay indicios de que esta terapia conduce a una mayor evidencia de Pseudomonas.
Pseudomonas aeruginosa: Con el aumento de la edad, aumenta la frecuencia de detección de Pseudomonas aeruginosa y otros gérmenes gramnegativos en las secreciones bronquiales. Aproximadamente el 30% de los pacientes de 18 años y el 75% de los de 45-49 años están crónicamente colonizados por este germen. Dado que no toda nueva detección de gérmenes va acompañada de un deterioro clínico, los frotis faríngeos o las muestras de esputo deben examinarse rutinariamente unas 4 veces al año. La detección de Pseudomonas aeruginosa requiere un largo periodo de incubación, por lo que debe garantizarse que el laboratorio microbiológico cumple los requisitos adecuados.
A la primera detección de Pseudomonas aeruginosa, está indicada la terapia antibiótica inhalada para su erradicación, independientemente de los síntomas clínicos. Para ello existen diferentes protocolos equivalentes:
- Tobramicina 2× 300 mg durante 4 semanas
- Colistina 2× 1 millón de UI durante 3 meses en combinación con ciprofloxacino 750 mg 2× 1 durante 3 semanas.
- Aztreonam lisado 3× 75 mg durante 4 semanas.
Se considera que la erradicación ha tenido éxito si 6 controles posteriores son negativos. Esto tiene éxito en aproximadamente el 80% de los casos.
Se dice que existe colonización crónica por Pseudomonas aeruginosa si más de la mitad de 6 controles en 12 meses son positivos. En este caso, se recomienda una terapia permanente de inhalación de antibióticos como tratamiento de supresión. La inhalación produce niveles de esputo más altos que la administración i.v. y tiene una toxicidad menor. La broncoconstricción tras la inhalación puede contrarrestarse con broncodilatadores. También existen inhalaciones en polvo. Aunque la inhalación provoca tos con más frecuencia que la inhalación húmeda, el menor tiempo de inhalación es una clara ventaja para muchos pacientes.
Existen los siguientes antibióticos inhalados:
Inhalación de polvo:
- Colistina 2× 1 cápsula dura de 125 mg en continuo
- Tobramicina 2× 4 cápsulas duras à 28 mg 28 días on/off
Inhalación húmeda:
- Aztreonam lisado 3× 75 mg. 28 días on/off
- Colistina 2× 1(-2) millones UI continua
- Levofloxacino 2× 240 mg 28 días on/off (a partir del 18º año)
- Tobramicina 2× 80/160/170/300 mg de forma continua o 28 días on/off
Exacerbación pulmonar
Una y otra vez, los pacientes experimentan exacerbaciones pumonales a pesar del tratamiento de supresión. Suelen caracterizarse por varios de los siguientes síntomas:
- Aumento de la tos
- aumento de la disnea
- Cambio en el volumen y color del esputo
- Fiebre
- Sensación de enfermedad, fatiga
- Pérdida de peso
- Caída del FEV1 en >10%
- Aumento de los cambios radiológicos
La exacerbación pulmonar puede tratarse intensificando la terapia inhalada, mediante terapia antibiótica oral o mediante terapia antibiótica intravenosa.
La ciprofloxacina y la levofloxacina están disponibles como fármacos orales activos contra las pseudomonas. A la hora de prescribirlo, debe tenerse en cuenta el riesgo de tendinitis o incluso de rotura del tendón.
La terapia intravenosa suele administrarse como terapia combinada durante 14 días. Los fármacos más utilizados son la ceftazidima, el meropenem y la piperacilina/tazobactam, que se combinan con un aminoglucósido, normalmente la tobramicina. En función del antibiograma, se pueden utilizar más antibióticos en caso de aumento de la resistencia y de falta de efecto de las terapias estándar.
Terapia antiinflamatoria
Las medidas de secretolisis y las terapias antibióticas conducen a una reducción de la inflamación. Otra terapia antiinflamatoria es el tratamiento con azitromicina. El antibiótico macrólido produce una mejora de la función pulmonar y una reducción de las exacerbaciones como terapia a largo plazo.
El tratamiento con altas dosis de ibuprofeno mostró efectos positivos en los ensayos clínicos, pero no se ha establecido en la práctica clínica debido a la preocupación por los efectos secundarios gastrointestinales o renales.
Los esteroides inhalados pueden formar parte de la terapia en pacientes con asma bronquial coexistente. El uso general en la fibrosis quística no está indicado. Lo mismo se aplica a la terapia con esteroides orales, que sólo se utiliza en pacientes con aspergilosis broncopulmonar alérgica (ABPA).
Trasplante de pulmón
En casos de deterioro grave de la función pulmonar, el trasplante de pulmón es una forma de mejorar la calidad de vida y el pronóstico a largo plazo. Debe considerarse una presentación en el centro de trasplantes en los siguientes casos:
- FEV1<30% o en caso de un rápido declive de la función pulmonar
- exacerbaciones pulmonares frecuentes, esp. para el tratamiento de cuidados intensivos
- Neumotórax recurrentes
- hemoptisis repetida
A nivel internacional, el 15% de los trasplantes de pulmón se realizan por fibrosis quística. Cabe suponer que esta cifra disminuirá significativamente debido a las mejores opciones terapéuticas que ofrecen los moduladores del CFTR.
Poliposis nasi y sinusitis crónica
Muchos pacientes con fibrosis quística padecen sinusitis crónica y poliposis nasi. La angustia causada por la obstrucción nasal, el dolor sobre las fosas nasales y la disminución de la capacidad olfativa suelen ser bastante importantes.
Como terapia conservadora y local, la irrigación nasal con NaCl al 0,9% para la eliminación de secreciones y costras está en primer lugar. También pueden utilizarse dispositivos de inhalación, algunos de los cuales también generan una vibración para lograr una mejor deposición en los senos paranasales. Los esteroides tópicos son especialmente eficaces para los pólipos nasales asociados a la alergia, y un ensayo de la terapia es apropiado en pacientes con fibrosis quística, aunque la inflamación neutrofílica suele ser la principal preocupación. Los α-simpaticomiméticos tópicos deben utilizarse con poca frecuencia debido a la taquifilaxia.
El efecto de los moduladores del CFTR sobre la poliposis nasi no se investigó en los estudios pivotales. Los estudios publicados entretanto demuestran que también se ha producido una reducción significativa de las quejas en este ámbito.
Si los síntomas no mejoran satisfactoriamente con la terapia conservadora, es posible el tratamiento quirúrgico. Sin embargo, la recidiva es muy frecuente tras la extirpación del pólipo únicamente.
Insuficiencia pancreática exocrina, pancreatitis recurrente
Aproximadamente el 85% de los pacientes con FQ padecen insuficiencia pancreática exocrina. El dolor abdominal resultante, las heces voluminosas y malolientes con depósitos grasos y la malnutrición son uno de los primeros síntomas de la enfermedad. La insuficiencia pancreática exocrina suele estar presente al nacer. La terapia consiste en la sustitución de las enzimas pancreáticas en las comidas.
La pancreatitis recurrente se produce en aproximadamente el 20% de los pacientes que no padecen insuficiencia pancreática exocrina. A menudo se trata de pacientes que tienen un curso más bien leve en términos de enfermedad pulmonar.
La terapia de la pancreatitis es sintomática. Los informes de casos individuales sugieren que los moduladores del CFTR pueden influir positivamente en la aparición de pancreatitis. Por otro lado, se ha descrito la aparición de pancreatitis tras el inicio de la terapia con moduladores de CFTR.
Complicaciones hepatobiliares
En cerca del 40% de los pacientes se desarrolla una fibrosis biliar focal, que progresa a cirrosis en cerca del 20% de estos pacientes. Las opciones terapéuticas son muy limitadas. El ácido ursodesoxicólico se recomienda en las directrices europeas, pero los datos son escasos. En casos concretos, puede ser necesario un trasplante de hígado. Aún se desconoce cómo influyen los moduladores del CFTR en el desarrollo de la fibrosis o la cirrosis.
Enfermedades malignas
Los pacientes con fibrosis quística tienen un mayor riesgo de padecer tumores gastrointestinales. Por ello, se recomienda la realización de una colonoscopia como examen preventivo a partir de los 40 años.
Diabetes mellitus
La prevalencia de la diabetes mellitus en la fibrosis quística aumenta significativamente con la edad y se sitúa en torno al 30% a los 30 años.
El tratamiento suele ser con insulina. Un ensayo clínico demostró que, al menos en los dos primeros años tras el diagnóstico, la terapia oral con repaglinida no es inferior a la terapia con insulina. No se puede evaluar de forma concluyente hasta qué punto el tratamiento con moduladores del CFTR influye en el desarrollo de la diabetes mellitus. También en este caso hay informes individuales positivos.
Infertilidad masculina
El 98% de los hombres con fibrosis quística son estériles debido a la aplasia bilateral congénita de los conductos deferentes (ABVDC) que se produce prenatalmente y provoca azoospermia. Dado que la producción de esperma suele estar preservada, los espermatozoides pueden obtenerse de los testículos o del epidídimo y utilizarse para la inseminación intrauterina (IIU), la fecundación in vitro (FIV) o la inyección intracitoplasmática de espermatozoides (ICSI).
Aspectos ginecológicos en pacientes con FQ
En las pacientes con fibrosis quística sólo se observa una ligera reducción de la fertilidad, debida, por ejemplo, a la mayor viscosidad de la secreción cervical. Numerosos informes de casos sugieren que la normalización de la viscosidad mediante el tratamiento con moduladores de CFTR conduce a una mejora de la fertilidad.
Debe aclararse en cada caso si debe continuarse el tratamiento con un modulador de CFTR en caso de embarazo. Los moduladores de CFTR atraviesan la barrera placentaria, pero no se han descrito efectos negativos en el niño. Los experimentos con animales tampoco mostraron indicios de daños.
Literatura:
- Ramsey BW, et al: A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365(18): 1663-1672.
- Volkova N, et al: Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. JCF 2020; 10: 68-70.
- Wainwright CE, et al: Tezacaftor-Ivacaftor en pacientes con fibrosis quística homocigotos para Phe508del CFTR. N Engl J Med 2015; 373(3): 220-231.
- Taylor-Cousar JL, et al: Tezacaftor-Ivacaftor en pacientes con fibrosis quística homocigotos para Phe508del. N Engl J Med 2017; 377(21): 2013-2023.
- Rowe SM, et al: Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med 2017; 377(21): 2024-2035.
- Middleton et al. Elexacaftor-Tezacaftor-Ivacftor para la fibrosis quística con un único alelo Phe508del. N Engl J Med 2019; 381(19): 1809-1819.
- Yang C, Montgomery M.: Dornasa alfo para la fibrosis quística. Cochrane Database Syst Rev 2021; 3(3): CD001127; doi: 10.1002/14651858.CD001127.pub5.
- Wark P, McDonald VM: Solución salina hipertónica nebulizada para la fibrosis quística. Cochrane Database Syst Rev 2018; 9(9): CD001506; doi: 10.1002/14651858.CD001506.pub4.
- Beswick DM, et al: Impacto de la terapia con reguladores de la conductancia transmembrana de la fibrosis quística en la rinosinusitis crónica y el estado de salud: análisis de tomografía computarizada de aprendizaje profundo y resultados comunicados por los pacientes. Ann Am Thorac Soc 2022; 19(1): 12-19.
- Hewer SCL, et al: Estrategias antibióticas para erradicar Pseudomonas aeruginosa en personas con fibrosis quística: Cochrane Database Syst Rev 2017; 4(4): CD004197.
InFo NEUMOLOGÍA Y ALERGOLOGÍA 2022; 4(4): 14-18









