La introducción de nuevos fármacos ha mejorado constantemente el pronóstico de los pacientes con mieloma múltiple en los últimos años. También se han ampliado los diagnósticos, en particular mediante análisis citogenéticos, que permiten una estratificación más precisa del riesgo. No obstante, la tasa de supervivencia a 5 años en el estadio III es sólo del 40%, por lo que aún hay margen para la innovación.
Como linfoma de células B con proliferación monoclonal de células plasmáticas en la médula ósea, el mieloma múltiple (MM) es una enfermedad extremadamente heterogénea. Aunque alrededor de una cuarta parte de los afectados son asintomáticos en el momento del diagnóstico, también hay cursos agudos con rápida destrucción ósea, disfunción renal pronunciada, fatiga, hipercalcemia y tendencia a la infección [1]. A medida que la población envejece, aumenta el número de nuevos casos, la mayoría de los cuales se producen entre los 70 y los 80 años. El mieloma múltiple sigue siendo una enfermedad rara, con una incidencia de unos 6 casos por cada 100.000 habitantes en hombres y 4 casos por cada 100.000 habitantes en mujeres, pero cabe esperar un aumento del número de casos en un tercio para 2040 debido únicamente al cambio en las estructuras de edad [2,3]. Por lo tanto, en el futuro seguirán aumentando los requisitos para una gestión eficaz con opciones terapéuticas efectivas y diagnósticos orientados a objetivos específicos.
Una mirada a la fisiopatología
En el mieloma múltiple, las células plasmáticas malignas producen inmunoglobulinas monoclonales completas o incompletas. Pueden detectarse como cadenas ligeras clonalmente aumentadas en suero y orina y también llevan el nombre de “paraproteína”. En la electroforesis de proteínas séricas, aparecen en forma del famoso “gradiente M”. La elevada concentración de estas inmunoglobulinas puede provocar diversos síntomas, como la amiloidosis AL, debido al depósito de proteínas mal plegadas. Además, puede producirse un síndrome de hiperviscosidad y -mucho más frecuentemente- un deterioro de la función renal. Esto se debe a que el aumento de cadenas ligeras se filtra glomerularmente y a menudo no puede reabsorberse por completo en los túbulos. Esto provoca la excreción en la orina, la llamada “proteinuria de Bence-Jones”. Además, las paraproteínas se acumulan en los glomérulos y precipitan en el túbulo distal al unirse a la proteína Tamm-Horsfall producida por las células epiteliales tubulares. Hallazgos: Glomeruloesclerosis y nefropatía por cilindros [4].
Otros síntomas del mieloma múltiple surgen principalmente del desplazamiento de la hematopoyesis normal y de la destrucción de los huesos. Estos mecanismos provocan dolor óseo, fracturas patológicas, hipercalcemia, anemia y susceptibilidad a las infecciones, entre otras cosas [1]. A menudo, las quejas de los afectados son inespecíficas, por lo que no es raro que pasen varios meses antes de que se realice un diagnóstico correcto.
La genética del mieloma múltiple es tan variada como su aspecto clínico. Las trisomías se encuentran en alrededor del 40% de los pacientes. También son frecuentes las translocaciones que afectan al locus de la cadena pesada de inmunoglobulina (IgH) en el cromosoma 14q32. Junto con las trisomías, pertenecen a las alteraciones genéticas primarias y pueden detectarse ya en las fases preliminares de la enfermedad [5]. A medida que la enfermedad progresa, se añaden aberraciones genéticas secundarias como el del(1p) o las mutaciones RAS, que configuran la clínica, la respuesta a la terapia y el pronóstico [3,6].
La conclusión es que la etiología del mieloma múltiple no está clara. La agrupación familiar es poco frecuente y los factores de riesgo siguen sin estar claros [3]. La enfermedad suele desarrollarse a partir de una gammapatía monoclonal de significado desconocido (GMSI) [7]. Si existe este estadio preliminar, el riesgo de progresión a mieloma múltiple u otra enfermedad linfoproliferativa que requiera tratamiento es de aproximadamente un 1% al año [8]. Sin embargo, no se ha establecido una detección precoz de los precursores, ya que esto aún no ha supuesto una mejora significativa del pronóstico [3].
Diagnóstico en transición
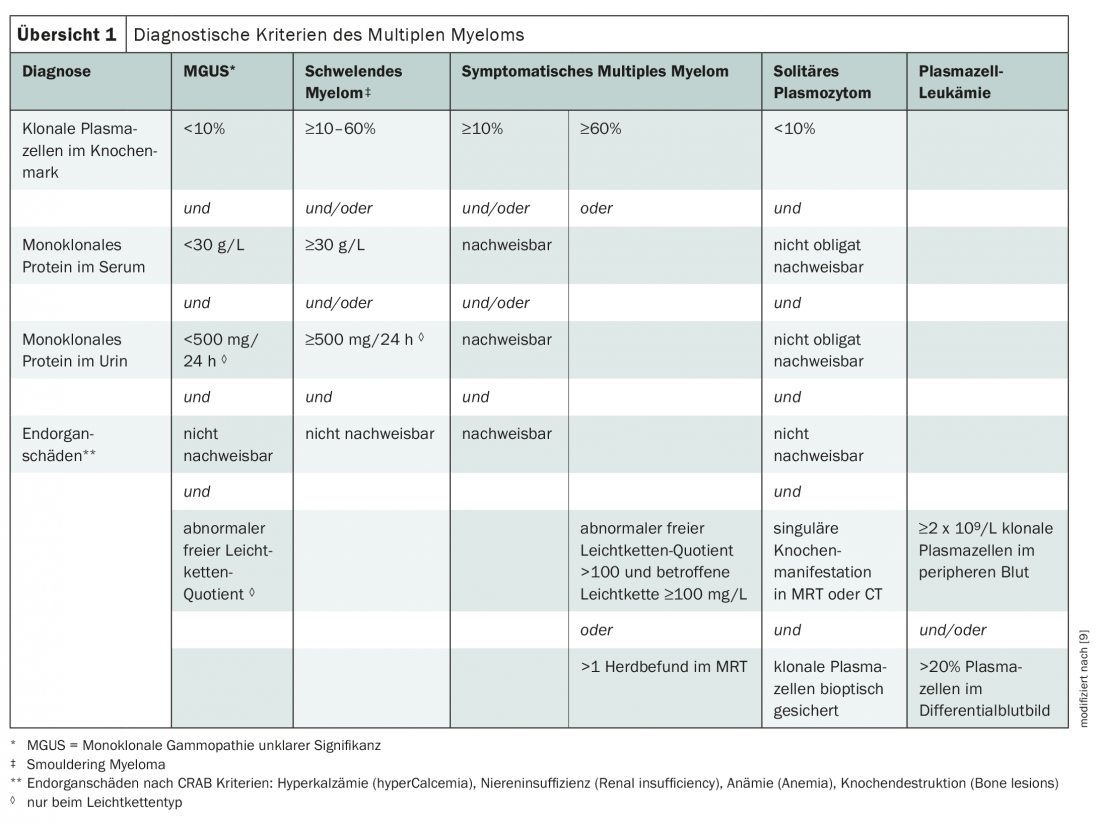
En la actualidad, para el diagnóstico del mieloma múltiple se utilizan principalmente los criterios del Grupo Internacional de Trabajo sobre el Mieloma (resumen 1) [3,9]. La enfermedad se clasifica según el tipo de paraproteína, siendo los mielomas IgG e IgA los más comunes. Si sólo se forman inmunoglobulinas incompletas, es decir, cadenas ligeras, se habla de “mieloma de cadenas ligeras”. Estos representan aproximadamente una quinta parte de los casos [3].
Además de la anamnesis, la exploración física, el hemograma y el laboratorio, una TC de cuerpo entero de dosis baja y una punción de médula ósea también forman parte del diagnóstico inicial. El diagnóstico por imagen se utiliza para detectar la osteólisis y la osteopenia [3]. En caso necesario, puede ampliarse con un examen de IRM para una diferenciación más precisa de los focos óseos y para el diagnóstico de las manifestaciones extramedulares. Esto puede ser especialmente útil para diferenciar el mieloma latente [10]. Las exploraciones FDG-PET también pueden proporcionar información sobre los focos extramedulares y la respuesta terapéutica, pero actualmente no forman parte de las investigaciones estándar [11]. Si se sospecha una afectación extramedular o si se presentan síntomas neurológicos, debe solicitarse una resonancia magnética de regiones específicas como muy tarde antes de iniciar la terapia. La ecocardiografía también está indicada si se sospecha de amiloidosis cardiaca [3].
Cada vez más, los diagnósticos genéticos desempeñan un papel importante en el tratamiento del mieloma múltiple. Los análisis sobre del(17p), t(4; 14) y t(14; 16) corresponden hoy en día al trabajo mínimo [3]. Estos cambios genéticos se asocian a un pronóstico significativamente peor. Otras aberraciones y también las puntuaciones pronósticas basadas en la expresión génica son relevantes desde el punto de vista del pronóstico, pero en la actualidad (todavía) no son predictivas de terapias específicas [6,12].
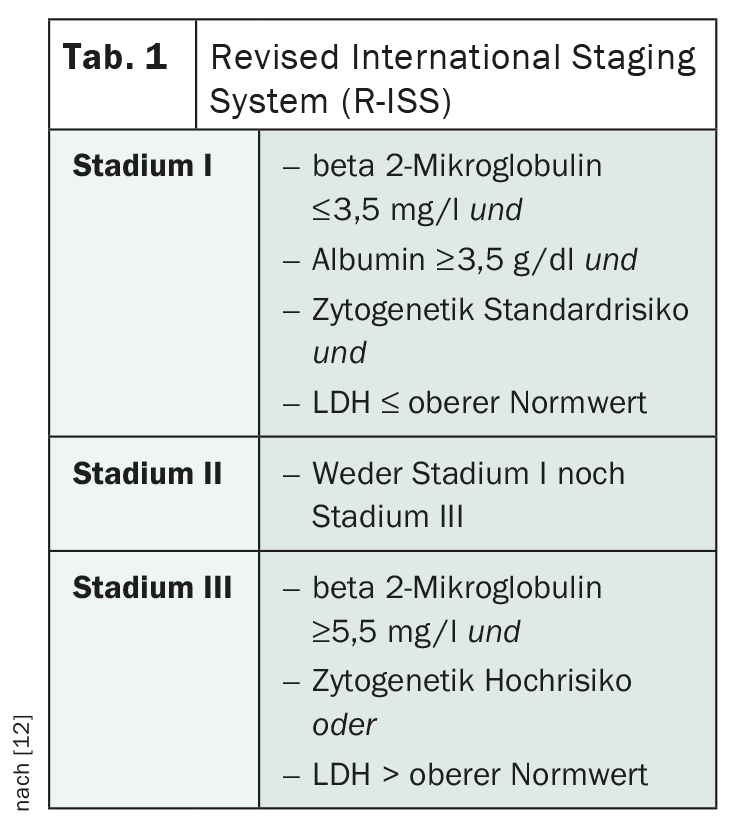
Clasificación para evaluar el pronóstico
La estadificación del mieloma múltiple desempeña un papel importante a la hora de estimar el pronóstico y evaluar la terapia más adecuada. La clasificación según Salmon y Durie, que se ha utilizado durante mucho tiempo, ha quedado obsoleta. En su lugar se utiliza el Sistema Internacional Revisado de Estadificación (R-ISS) del Grupo Internacional de Trabajo sobre el Mieloma (IMWG) (Tab. 1) [12]. Basándose en esta estadificación, los individuos afectados se dividen en tres grupos pronósticos, teniendo en cuenta la albúmina sérica, la beta 2-microglobulina, la LDH y las aberraciones citogenéticas.
La presencia de enfermedad residual mínima (ERM ) tras la terapia también tiene importancia pronóstica. Está presente en la gran mayoría de los pacientes incluso después de alcanzar una remisión completa según los criterios actuales y se correlaciona con un peor pronóstico [13]. Dado que los análisis de ERM aún no tienen ningún valor predictivo para la terapia posterior, actualmente no son un estándar en los exámenes de seguimiento [3].
La terapia de primera línea de un vistazo
Además de los cursos sintomáticos y los denominados criterios CRAB (hipercalcemia – C, insuficiencia renal – R, anemia – A y afectación ósea – B), también existen otros parámetros radiológicos y serológicos que, según los criterios del IMWG, representan indicaciones para el inicio de la terapia. Entre ellos se incluyen la evidencia de biomarcadores definitorios de mieloma, un contenido de células plasmáticas clonales en la médula ósea de >60%, un cociente de cadenas ligeras libres en suero de >100 (cadena ligera afectada/no afectada) y lesiones focales de más de 1 cm en la IRM [3].
Si existe una indicación de terapia, se distingue principalmente entre los pacientes aptos para un trasplante de células madre y los que deben ser tratados sin terapia de dosis altas debido a comorbilidades. Si el trasplante no es una opción, pueden utilizarse diversas combinaciones de dos y tres fármacos en el tratamiento farmacológico. Los pacientes afectados con insuficiencia renal, alta actividad de la enfermedad y citogenética desfavorable deben recibir preferentemente terapia con bortezomib como componente [3]. Otros agentes que se utilizan son la ciclofosfamida, el melfalán, la lenalidomida y los esteroides. Desgraciadamente, sólo se compararon directamente algunos de los esquemas comunes. En general, las combinaciones de tres sustancias con un inhibidor del proteasoma, un inmunomodulador y un esteroide parecen ser superiores a las combinaciones duales. Por lo tanto, si el estado general es bueno, son preferibles [3].
Si se dispone de un trasplante autólogo de células madre, es el tratamiento de elección en la primera línea terapéutica. Hasta ahora, ningún nuevo fármaco ha podido igualar al trasplante en cuanto a tasa de remisión, profundidad de la respuesta y supervivencia sin progresión [14,15]. Los efectos adversos de la terapia con dosis altas son un factor limitante para la indicación del trasplante autólogo de células madre. Esto requiere una buena función orgánica y la ausencia de comorbilidades significativas [16]. Hace sólo unos años, se recomendaba la terapia de inducción con vincristina-antraciclina. Desde entonces, esto se ha sustituido por terapias combinadas con los nuevos fármacos talidomida, bortezomib y lenalidomida, que dan lugar a tasas de respuesta significativamente mejores. También en este caso, los datos sobre la comparación directa de las diferentes terapias combinadas son desgraciadamente limitados; las remisiones completas se alcanzan en un 20-40% de los casos [3]. En la actualidad, la elección de la terapia de inducción se basa principalmente en factores individuales del paciente, los efectos secundarios y la disponibilidad del fármaco.
El trasplante de células madre tras la terapia de inducción puede realizarse como trasplante único o en tándem. En este último caso, se realiza un segundo trasplante autólogo en un plazo de seis meses. Esto conduce a una prolongación de la supervivencia libre de progresión, y en los pacientes en estadio III R-ISS se observó una mayor supervivencia global además de una mayor supervivencia libre de progresión [17]. Sin embargo, también debe tenerse en cuenta el aumento de la toxicidad de una segunda terapia con dosis altas. Actualmente, se recomienda el trasplante en tándem para los pacientes del grupo III de R-ISS y para aquellos con citogenética de alto riesgo, teniendo en cuenta diversos análisis de subgrupos y a largo plazo [3]. Por supuesto, debe garantizarse la presencia de un número suficiente de células madre autólogas conservadas. En la actualidad, se utiliza melfalán 200 mg/m2 para la terapia de dosis alta [18]. Los regímenes de acondicionamiento alternativos que utilizan ciclofosfamida y/o irradiación corporal total ya no se recomiendan debido a su mayor toxicidad. La terapia óptima con dosis altas también sigue siendo actualmente objeto de diversos estudios; una administración adicional de busulfán no condujo a una prolongación de la supervivencia global. Por desgracia, la adición de bortezomib tampoco resultó eficaz [19]. No existen datos concluyentes sobre la aplicación de una terapia de consolidación tras un trasplante autólogo de células madre; ésta puede ser especialmente útil en pacientes con una respuesta subóptima tras el trasplante o con una citogenética de alto riesgo [20].
Las recomendaciones para la terapia de mantenimiento son más claras. Tras años de no recomendarse, ahora es una parte importante del estándar terapéutico, gracias a los nuevos fármacos. Así, el bortezomib o la lenalidomida se utilizan en el grupo de alto riesgo y la lenalidomida en el de riesgo estándar. Y lo mismo ocurre con la respuesta completa [3,19,21].
Un factor terapéutico importante que no debe olvidarse, independientemente del estado del trasplante, es la osteoprotección. Especialmente en casos de afectación ósea y durante la terapia con glucocorticoides, deben utilizarse bifosfonatos o -sobre todo en casos de insuficiencia renal- denosumab para prevenir mejor la pérdida ósea. El zolendronato se considera el bifosfonato de primera elección para el mieloma múltiple [3].
¿Y en caso de progresión o recidiva?
Las progresiones y recaídas tras la terapia de primera línea constituyen un reto importante en el mieloma múltiple. Entre otras cosas, porque la población de pacientes es aún más heterogénea con las líneas de terapia posteriores. Las personas afectadas han recibido diferentes tratamientos y han tenido diferentes experiencias. La elección de los fármacos se basa, entre otras cosas, en la eficacia y la tolerabilidad del tratamiento de primera línea. Así, si la experiencia de primera línea es buena, se puede utilizar un fármaco de la misma clase de sustancia en el tratamiento de segunda línea. Sin embargo, en caso de baja eficacia o mala tolerabilidad, está indicado un cambio de clase de fármaco. En la actualidad, hay abundancia de nuevos fármacos y combinaciones de fármacos. En función de la presentación clínica, las terapias previas y las comorbilidades, puede seleccionarse así una amplia gama de sustancias y secuencias [3].
Mientras que las recaídas tempranas y tardías se tratan preferentemente con un trasplante de células madre, existen diversos regímenes terapéuticos para el resto de recaídas y para las contraindicaciones del trasplante. Éstos se basan en inhibidores del proteasoma o en inmunomoduladores y constan de dos o tres sustancias activas (resumen 2) . En este caso también se utiliza el anticuerpo anti-CD38 daratumumab. En general, las combinaciones triples son más eficaces que las dobles, pero también hay que tener en cuenta más toxicidades [3,19].

Con la creciente investigación de nuevos agentes activos para la terapia del mieloma múltiple, se han logrado algunos éxitos en los últimos años, sobre todo en la terapia de mantenimiento y en las líneas de tratamiento avanzadas. Sin embargo, el pronóstico en el grupo de alto riesgo y en la enfermedad recurrente sigue siendo desfavorable en la actualidad. Gracias al análisis citogenético cada vez más extendido, ya es posible una mejor estratificación del riesgo. Esto hace albergar esperanzas de que en el futuro se disponga no sólo de opciones diagnósticas, sino potencialmente de opciones terapéuticas más específicas.
Literatura:
- Kyle RA, et al: Revisión de 1027 pacientes con mieloma múltiple recién diagnosticado. Mayo Clin Proc. 2003; 78(1): 21-33.
- NICER: Estadísticas nacionales sobre la incidencia del cáncer. www.nicer.org/en/statistics-atlas/cancer-incidence (último acceso 27.02.2021)
- Wörmann B, et al: Mieloma múltiple. Guía Onkopedia. www.onkopedia.com/de/onkopedia/guidelines/multiples-myelom/@@guideline/html/index.html (último acceso 27.02.2021)
- Dimopoulos MA, et al: Patogenia y tratamiento de la insuficiencia renal en el mieloma múltiple. Leucemia. 2008; 22(8): 1485-1493.
- Mikulasova A, et al.: El espectro de mutaciones somáticas en la gammapatía monoclonal de significado indeterminado indica un paisaje genómico menos complejo que el del mieloma múltiple. Haematologica. 2017; 102(9): 1617-1625.
- Bergsagel PL, Chesi MV: Clasificación molecular y estratificación del riesgo del mieloma. Hematol Oncol. 2013; 31 Suppl 1(0 1): 38-41.
- Landgren O, et al.: La gammapatía monoclonal de significado indeterminado (GMSI) precede sistemáticamente al mieloma múltiple: un estudio prospectivo. Sangre. 2009; 113(22): 5412-5417.
- Dispenzieri A, et al: Prevalencia y riesgo de progresión de la gammapatía monoclonal de cadena ligera de significado indeterminado: un estudio de cohortes retrospectivo basado en la población. Lancet. 2010; 375(9727): 1721-1728.
- Rajkumar SV, et al: Criterios actualizados del Grupo Internacional de Trabajo sobre el Mieloma para el diagnóstico del mieloma múltiple. Lancet Oncol. 2014; 15(12): e538-548.
- Dimopoulos MA, et al: Papel de la resonancia magnética en el tratamiento de pacientes con mieloma múltiple: declaración de consenso. J Clin Oncol. 2015; 33(6): 657-664.
- Cavo M, et al: Papel de la (18)F-FDG PET/TC en el diagnóstico y tratamiento del mieloma múltiple y otros trastornos de células plasmáticas: declaración de consenso del Grupo Internacional de Trabajo sobre el Mieloma. Lancet Oncol. 2017; 18(4): e206-e17.
- Palumbo A, et al: Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol. 2015; 33(26): 2863-2869.
- Munshi NC, et al: Asociación de la enfermedad mínima residual con resultados de supervivencia superiores en pacientes con mieloma múltiple: un metaanálisis. JAMA Oncol. 2017; 3(1): 28-35.
- Barlogie B, et al.: Superioridad del trasplante autólogo en tándem sobre la terapia estándar para el mieloma múltiple no tratado previamente. Sangre. 1997; 89(3): 789-793.
- Gay F, et al: Quimioterapia más lenalidomida frente a trasplante autólogo, seguido de lenalidomida más prednisona frente a mantenimiento con lenalidomida, en pacientes con mieloma múltiple: un ensayo aleatorizado, multicéntrico, de fase 3. Lancet Oncol. 2015; 16(16): 1617-1629.
- Merz M, et al: Supervivencia de los pacientes ancianos con mieloma múltiple: efecto del trasplante autólogo de células madre por adelantado. Eur J Cancer. 2016; 62: 1-8.
- Cavo M, et al: El doble autotrasplante de células madre prolonga significativamente la supervivencia libre de progresión y la supervivencia global en comparación con el autotrasplante único en el mieloma múltiple recién diagnosticado: un análisis del estudio de fase 3 EMN02/HO95. Sangre. 2017; 130 (suplemento 1): 401.
- Giralt S: 200 mg/m(2) de melfalán: el patrón oro para el mieloma múltiple. Nat Rev Clin Oncol. 2010; 7: 490-491.
- Dimopoulos MA, et al: Mieloma múltiple: Guía de práctica clínica de la EHA-ESMO para el diagnóstico, tratamiento y seguimiento. Anales de Oncología. 2021; 32(3): 309-322.
- Einsele H, et al: La consolidación adaptada a la respuesta con bortezomib tras el TACS mejora la supervivencia libre de progresión en el mieloma múltiple de diagnóstico reciente. Leucemia. 2017; 31(6): 1463-1466.
- McCarthy PL, et al: Mantenimiento con lenalidomida tras trasplante autólogo de células madre en mieloma múltiple recién diagnosticado: un metaanálisis. J Clin Oncol. 2017; 35(29): 3279-3289.
InFo ONCOLOGÍA Y HEMATOLOGÍA 2021; 9(2): 22-26