
¿Cómo pueden utilizarse con sensatez los inhibidores de la cinasa? ¿Cuáles son las ventajas o desventajas de una combinación? ¿Cómo puede contribuir la secuenciación genómica a un enfoque terapéutico individualizado con dichos inhibidores y qué diseños de estudio deberían utilizarse para probarlo? ¿Qué se puede aprender de los estudios fallidos y de los procesos de desarrollo de los inhibidores de la cinasa conocidos? Todas estas cuestiones se debatieron en el Congreso de la ESMO celebrado en Ámsterdam.
Annette K. Larsen, de París, planteó la cuestión de si combinar inhibidores de la cinasa y cómo hacerlo:
“El desarrollo de nuevos agentes anticancerígenos dirigidos a las vías de señalización oncogénica representa un gran avance conceptual. Sin embargo, los resultados clínicos a menudo no han estado a la altura de las expectativas, en parte debido a mutaciones aguas abajo, a bucles de retroalimentación inesperados o a las denominadas interacciones cruzadas de las vías de señalización.”
Por este motivo, hoy en día se presta mucha atención a la selección de varias rutas de señal simultáneamente o de diferentes pasos en la misma ruta de señal. A menudo, los inhibidores de la cinasa se añadían directamente a los agentes citotóxicos establecidos sin ajustar la dosis, lo que a veces provocaba graves efectos secundarios tóxicos. En su presentación, Larsen expuso las posibilidades y limitaciones de las combinaciones de inhibidores de la cinasa.
Dos ensayos recientes de fase III (PACCE y CAIRO2) evaluaron la adición de anticuerpos monoclonales (mAbs) dirigidos contra el EGFR, cetuximab y panitumumab, respectivamente, al bevacizumab más quimioterapia en pacientes con cáncer colorrectal (CCR). Esta combinación no sólo se asoció a una menor supervivencia libre de progresión, sino también a una peor calidad de vida, también en pacientes con un tumor KRAS de tipo salvaje.
“¿Por qué no funcionó la combinación de los dos mAbs? En primer lugar, la combinación de agentes diana era demasiado tóxica y, en segundo lugar, no es activa, es decir, ambos inhiben los ligandos o receptores extracelulares, pero tienen efectos limitados, quizá incluso nulos, sobre la señalización de la tirosina quinasa receptora (RTK)”, afirma Larsen. Concluye que, aunque la combinación de agentes dirigidos contra el VEGF y el EGFR es posible, no tiene por qué utilizarse necesariamente al mismo tiempo que la quimioterapia. El estado de mutación de KRAS desempeña un papel aquí, especialmente en los mAbs dirigidos al EGFR.
Enfoques terapéuticos individualizados
El Prof. Dr. med. Emile E. Voest, de Utrecht, habló sobre las posibilidades de incluir la secuenciación genómica en la toma de decisiones terapéuticas: “La información detallada sobre cómo está formado genéticamente un tumor permite una selección más selectiva de pacientes concretos para una terapia respectiva. Ejemplos como el trastuzumab para la expresión HER2 en el cáncer de mama, el imatinib para las translocaciones BCR-ABL en la leucemia, el vemurafenib y el crizotinib contra las mutaciones V600E en el melanoma o las translocaciones ALK-EML4 en el cáncer de pulmón han demostrado claramente la validez del concepto. Estos éxitos espectaculares se ven empañados por el hecho de que son temporales porque a menudo se desarrolla resistencia”.
Gracias a los avances en la tecnología de secuenciación, ahora es posible generar información detallada sobre las anomalías genómicas de un tumor. Los análisis de un solo gen (es decir, BRAF, KRAS) serán sustituidos por análisis de todo el genoma en un futuro próximo. Este enfoque global permite obtener información a nivel de las vías de señalización en lugar de a nivel de genes individuales. “Por ejemplo, no pudimos encontrar una correlación entre el éxito de la quimioterapia y las mutaciones específicamente rastreables en el gen PI3K en pacientes con cáncer de mama, pero sí con las mutaciones en la vía de señalización PI3K”, afirma el Prof. Voest.
La heterogeneidad tumoral es una dimensión importante del crecimiento tumoral y a menudo se produce el crecimiento clonal de poblaciones resistentes durante el tratamiento. En este caso, los métodos de “secuenciación (ultra)profunda” pueden ayudar a detectar e identificar los clones de forma precoz, permitiendo un enfoque de tratamiento anticipado. La opinión actual es que será más fácil encontrar un perfil genético predictivo para los fármacos con un mecanismo de acción específico (como el vemurafenib) que para los inhibidores de la quinasa de amplio espectro (como el sunitinib). Teniendo en cuenta los hallazgos sobre la mencionada correlación entre el éxito de la quimioterapia y la vía de señalización PI3K, este planteamiento puede exponerse como un malentendido.
“Estamos al principio de una era en la que las pruebas genéticas exhaustivas del tumor y del ADN de la línea germinal formarán parte del diagnóstico habitual en los pacientes con cáncer. En particular, la interpretación competente de los datos extremadamente complejos será crucial para la correcta elección de la terapia guiada por el ADN. Para ello, se necesitan grandes bases de datos que ofrezcan la posibilidad de vincular el éxito clínico con los datos genéticos”, concluyó su conferencia el Prof. Voest.
Estudios del genotipo frente a la cesta
“Los estudios de genotipo se basan en someter a un número determinado de pacientes con una enfermedad a pruebas para determinar su genotipo y luego asignarles distintos fármacos dirigidos en función de su perfil”, afirma el Dr. José Baselga, de Nueva York (Fig. 1). “Los problemas de este enfoque son los siguientes: Los fármacos utilizados en cada grupo no suelen ser los mejores de su clase, pero están disponibles (por ejemplo, el primer ensayo BATTLE utilizó sorafenib como inhibidor del RAF). Además, si se utiliza la aleatorización, puede parecer poco ético durante el transcurso del ensayo aleatorizar a algunos pacientes que no serán tratados según su perfil. Además, el número total de participantes suele ser bajo, por lo que este diseño puede adquirir a menudo muy pocos pacientes con mutaciones raras para comparar de forma válida el resultado clínico con el perfil genético (por ejemplo, BRAF en el cáncer de pulmón o mutaciones ERBB2 en el cáncer de ovario).”
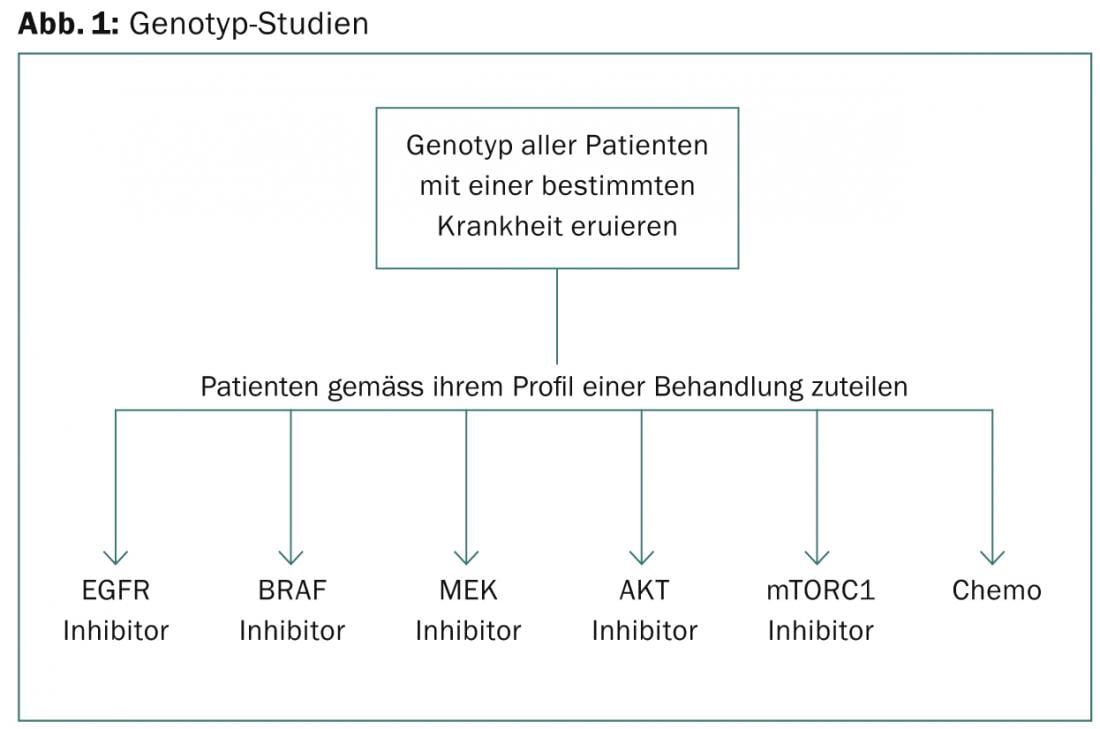
Los llamados. Los estudios sobre cestas, en cambio, permiten probar una hipótesis concreta, por ejemplo: ¿Responden al vemurafenib los pacientes con cáncer biliar cuyo tumor alberga mutaciones BRAF(V600E)? Además, como este tipo de estudio se basa en una colección de cohortes de enfermedades específicas, puede utilizarse para investigar la influencia de la ascendencia en la respuesta a los fármacos. Además, el muestreo de tejidos puede explicar la heterogeneidad de la respuesta. La principal crítica a los estudios en cesta es que pueden pasar por alto a aquellos pacientes que podrían responder pero no tienen el biomarcador que buscan. Además, la identificación de los participantes sigue siendo un obstáculo: según Baselga, es por tanto imperativo separar el protocolo de cribado del protocolo de tratamiento.
Aprender de los errores
¿Cómo podemos aprender de los ensayos fallidos con inhibidores de la cinasa? Esta pregunta fue planteada por el Dr. Stefan Sleijfer, de Rotterdam: “En los últimos años, varios nuevos inhibidores de la cinasa se aprobaron con rapidez y éxito gracias a los resultados de estudios en parte sensacionales. Sin embargo, muchos de estos agentes también fracasaron, a veces ya en la fase clínica inicial, y otras veces sólo después de que se hubieran llevado a cabo extensos y costosos estudios de fase III. Tanto de los procesos de desarrollo exitosos como de los fallidos pueden extraerse conclusiones valiosas para futuros diseños de estudios.”
Los inhibidores de la cinasa que tienen más éxito son los que se dirigen directamente al producto de un gen mutado y lo inhiben, es decir, afectan a un subconjunto específico de una enfermedad. Una vez que se conoció el mecanismo de acción de estos fármacos y, por tanto, se pudo seleccionar específicamente a los pacientes aptos, las sustancias activas se probaron clínicamente en grupos de pacientes específicamente seleccionados desde el principio. Algunos ejemplos de éxito son el imatinib para los tumores malignos del estroma gastrointestinal (GIST), el vemurafenib para el melanoma con mutación BRAF(V600E) o el crizotinib para el cáncer de pulmón no microcítico (CPNM) con gen de fusión EML4-ALK.
Una importante característica común de aquellos fármacos que fracasaron o sólo obtuvieron la aprobación tras un arduo y costoso camino es que el mecanismo de acción era desconocido antes de que comenzaran los ensayos clínicos. Por lo tanto, no se pudieron establecer perfiles predictivos, no se pudo identificar a priori a los pacientes con probabilidades de responder y los ensayos clínicos se realizaron en poblaciones de pacientes no seleccionadas. Ejemplos actuales son los antagonistas de IGF-1R y los inhibidores de mTOR.
“Así que la lección más importante de estos estudios es el beneficio y la necesidad de descubrir el mecanismo de acción exacto del fármaco investigado en entornos preclínicos y de identificar marcadores predictivos antes de iniciar grandes ensayos clínicos. Si se desconoce el mecanismo, sigue siendo cuestionable si el fármaco en cuestión debe entrar en el proceso de desarrollo. Sobre todo porque esto requiere una cantidad extremadamente grande de recursos y dinero que podría emplearse mejor en otros compuestos que deben probarse. Si, a pesar de todo, ya se está desarrollando un fármaco con un mecanismo de acción desconocido, pueden utilizarse diseños adaptativos. Por ejemplo, la recogida de biomaterial es entonces esencial para poder descubrir retrospectivamente perfiles predictivos, como ocurrió con los inhibidores de la quinasa EGFR en el CPNM”, resumió el Dr. Sleijfer.
Fuente: “Optimal use of Targeted Kinase Inhibitors (TKIs)”, Simposio en el Congreso de la ESMO, 27 de septiembre – 1 de octubre de 2013, Ámsterdam.
InFo ONCOLOGÍA Y HEMATOLOGÍA 2014; 2(1): 45-47









