As doenças raras na Suíça são definidas por uma prevalência inferior a 1:2000**. Existem mais de 1000 doenças de pele raras reconhecidas em dermatologia. A rede “RareSkinCH” liga centros de referência e associações de doentes com o objetivo de melhorar a situação dos cuidados prestados às pessoas afectadas por doenças cutâneas raras.
A rede “RareSkinCH” é uma comissão do SGDV e faz parte do “Kosek”, o centro de coordenação nacional para as doenças raras [1]. A iniciativa “RareSkinCH” promove a colaboração entre hospitais, profissionais de saúde e associações de doentes [2]. O objetivo é otimizar a gestão global dos doentes com doenças cutâneas raras. A rede esforça-se por atingir os seguintes objectivos
- diagnóstico correto e precoce,
- Acesso a testes de diagnóstico adequados,
- cuidados médicos em centros com competências alargadas,
- Acesso a novas terapias, reabilitação e cuidados de apoio e psicossociais.
Os grupos mais importantes de doenças cutâneas raras incluem Ictioses, queratodermia palmo-plantar, displasia ectodérmica, epidermólise bolhosa, anomalias pigmentares (por exemplo, albinismo), anomalias vasculares (por exemplo, linfedema), nevos congénitos, doenças do tecido conjuntivo, doenças tumorais raras (por exemplo, linfomas cutâneos), certas doenças auto-imunes, alguns síndromes auto-inflamatórios e reacções cutâneas tóxico-imunológicas graves [2].
** FOPH: Conceito nacional para as doenças raras. www.bag.admin.ch/bag/de/home/strategie-und-politik/ politische-auftraege-und-aktionsplaene/nationales-konzeptseltene-krankheiten.htm, (último acesso em 05.06.2024).
Exemplos da prática clínica quotidiana
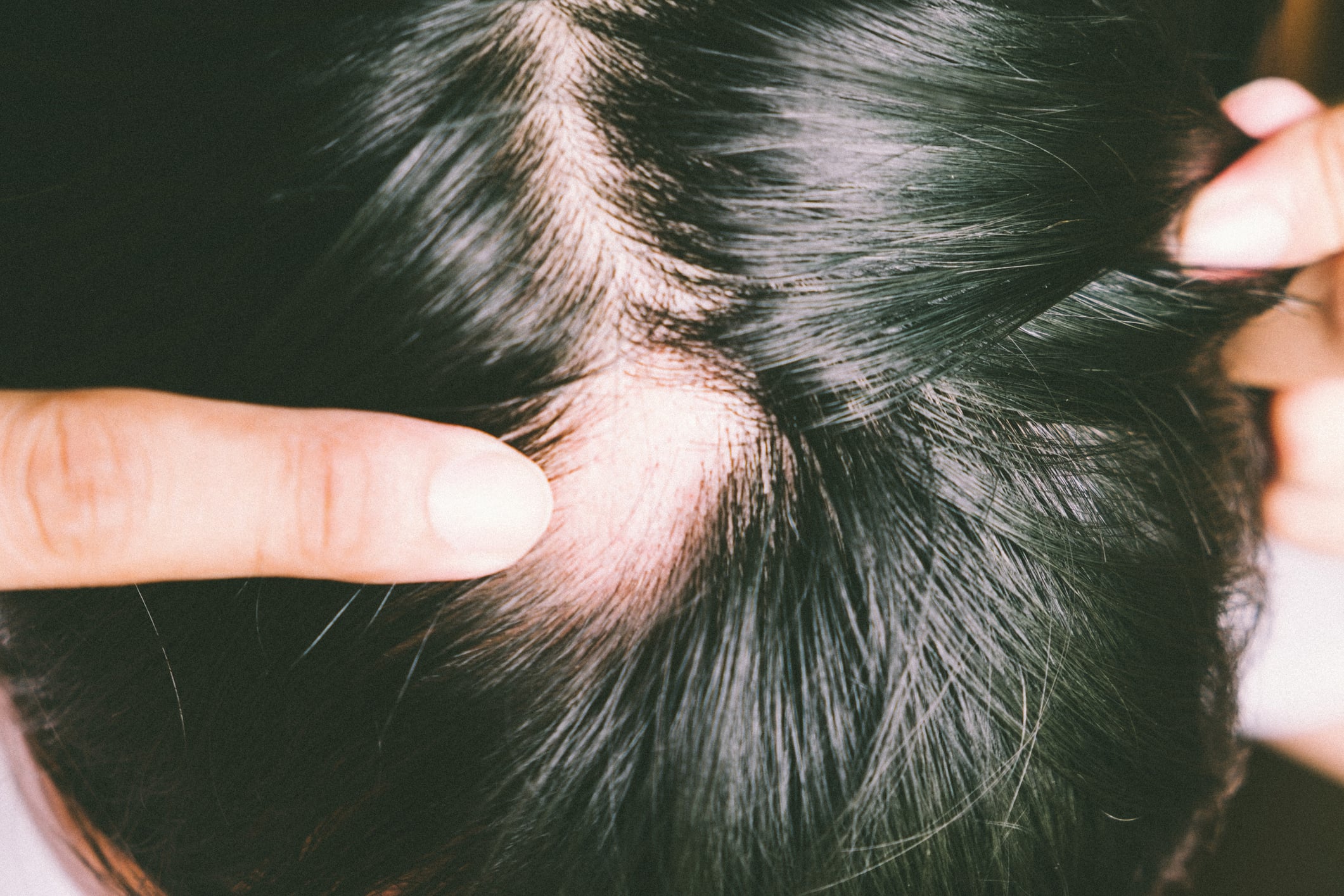
O grupo das doenças da queratinização é muito heterogéneo e pode ser dividido em cerca de 100 doenças. Estas incluem queratoses palmoplantares, ictioses e queratoses que, ao contrário das queratoses palmoplantares, não se limitam exclusivamente à pele das virilhas, das palmas das mãos e das plantas dos pés e podem apresentar-se como hiperqueratose ou disqueratose. A eritroqueratodermia, que se caracteriza por focos eritematosos e escamosos, é também uma doença da queratinização [3].
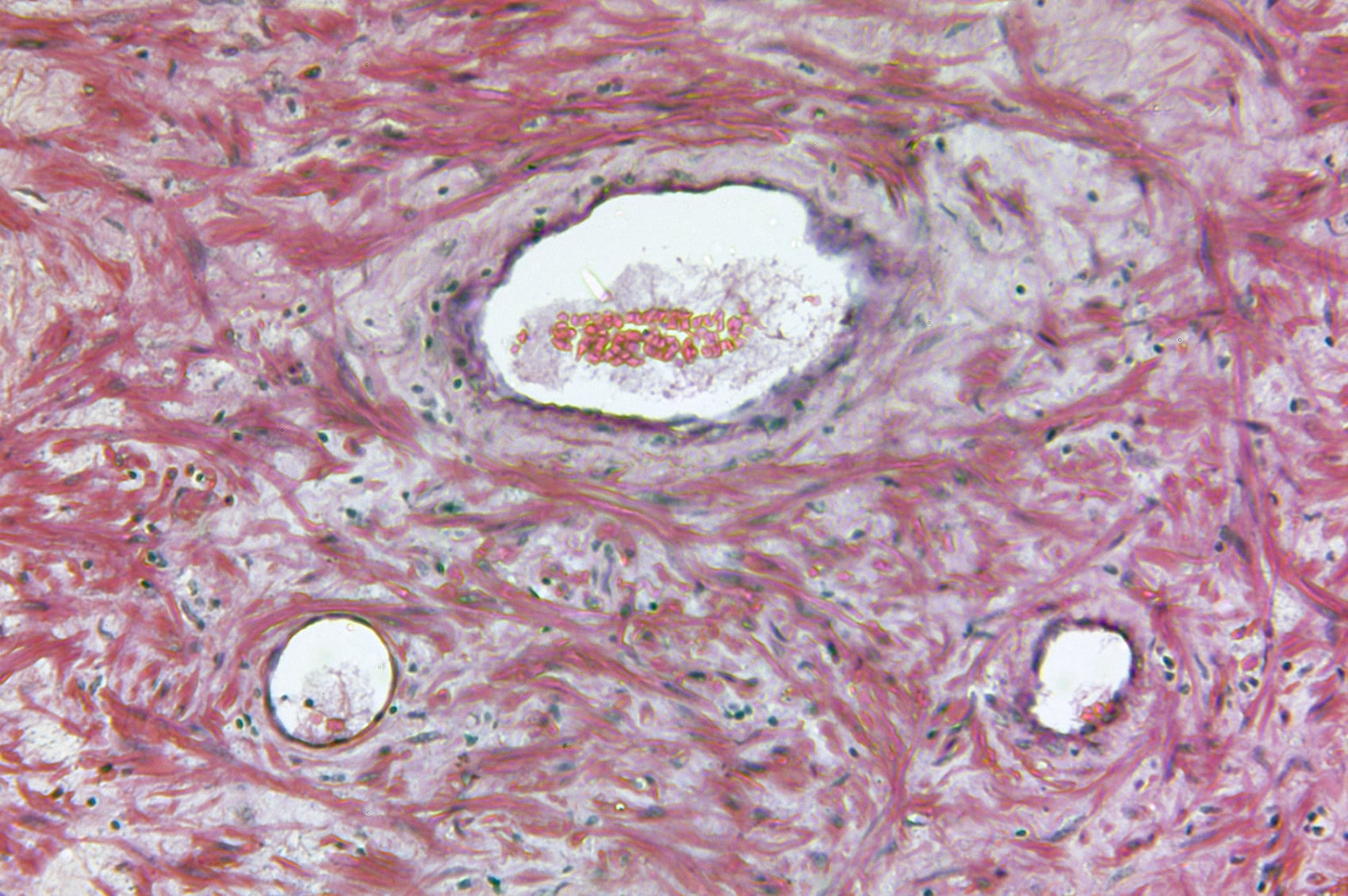
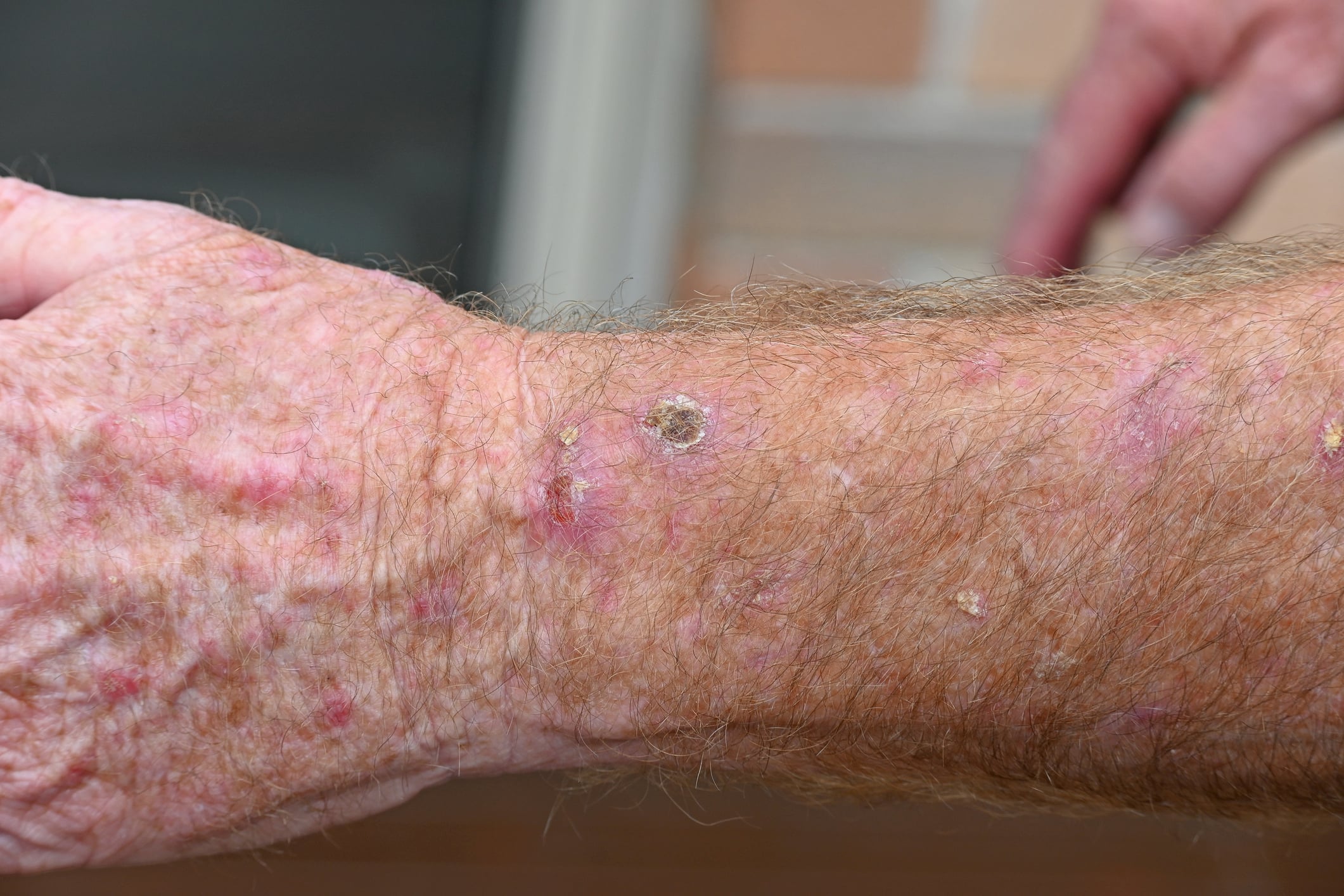
Doente do sexo feminino, 32 anos de idade, com queratose epidermolítica palmoplantar hereditária: A doente apresentou-se na policlínica com placas hiperqueratóticas dolorosas e rágades nas palmas das mãos e plantas dos pés [6]. A história familiar era positiva. Na região palmo-plantar, havia hiperqueratoses estriadas, amareladas a acastanhadas, nas áreas expostas à pressão. Os testes genéticos revelaram uma variante patogénica heterozigótica não descrita anteriormente no exão 4 da desmogleína-1 (DSG1) [c.349C>T, p.(Arg117*)]. A histopatologia revelou orto-hiperqueratose e acantose irregular com um estrato granuloso pronunciado e acantólise. A microscopia eletrónica mostrou queratinócitos basais claramente alongados e numerosos tonofilamentos. O DSG1 é expresso principalmente nas camadas suprabasais da epiderme e da mucosa e é essencial para a adesão intercelular dos desmossomas. Mutações isoladas no DSG1 levam a acantólise com hiperqueratose consecutiva nas palmas das mãos e plantas dos pés, particularmente em áreas sujeitas a stress mecânico. As opções de tratamento consistem em queratólise não mecânica e alívio da pressão. Os retinóides sistémicos devem ser utilizados com precaução nas queratoses palmo-plantares epidermolíticas, uma vez que os sintomas podem agravar-se durante o tratamento.
| Factos importantes sobre as dermatoses raras |
| As dermatoses raras mono e poligénicas representam um grupo significativo de doenças com mais de 1000 entidades [8]. Nas últimas duas décadas, a sua investigação tornou-se um foco importante na dermatologia [9]. |
| [10,11]Mais de 70% das doenças raras são causadas por defeitos num único gene . |
| [8,11]Há um número crescente de registos e redes em que são recolhidos dados de doentes com doenças raras. A nível europeu, estas incluem o projeto Biomed2 Geneskin da UE, a Orphanet e a ERN Skin (Rede Europeia de Referência sobre Doenças de Pele Raras e Não Diagnosticadas). |
| As genodermatoses causadas por mutações sem sentido podem agora ser tratadas com agentes indutores de leitura, como os aminoglicosídeos [9]. Em contrapartida, as genodermatoses associadas a variantes de ganho de função podem ser tratadas através do bloqueio das vias de sinalização activadas [8]. |
As mucinoses são outro grupo heterogéneo de doenças raras. Trata-se de dermatoses de depósito cuja caraterística comum é o aumento patológico da mucina, uma mistura gelatinosa de glicosaminoglicanos ácidos, no tecido conjuntivo. A consistência gelatinosa resulta da propriedade especial dos glicosaminoglicanos ácidos de se ligarem mais de 1000 vezes o seu próprio peso em água. [4,5]As cinco principais mucinoses são o mixoedema generalizado, o mixoedema pré-tibial, o líquen mixoedematoso, a mucinose eritematosa reticular e o escleredema .
Doente de 52 anos com escleromixedema (síndroma de Arndt-Gottron): O doente apresentou-se na consulta externa devido a pápulas disseminadas que estavam presentes há um ano [7]. O tratamento anterior com acitretina não tinha revelado qualquer melhoria dos resultados. Não eram conhecidas doenças crónicas. Os sintomas B não estavam presentes. O doente referiu um abuso de nicotina de 30 anos-maço. Clinicamente, foram observadas massas de pápulas lenticulares com um aspeto ceroso no pescoço, nos lados extensores dos braços e nas costas das mãos, bem como nos lados extensores dos dedos. Histologicamente, foram observadas acumulações intersticiais de mucina rodeadas por fibroblastos na derme superior a profunda. Foram encontrados infiltrados linfocíticos perivasculares subtis. Outros diagnósticos revelaram uma gamopatia monoclonal IgG tipo Lambda. Do ponto de vista hematológico, não havia inicialmente qualquer necessidade de atuar a este respeito. A radiografia do tórax e a ecografia do abdómen não revelaram quaisquer anomalias. Foi iniciada terapêutica sistémica com isotretinoína 20 mg durante 6 meses. Durante o tratamento, os achados locais progrediram com alterações cutâneas crescentes na área do tronco e, entretanto, o envolvimento da glabela e das sobrancelhas também se tornou evidente. Por conseguinte, foi decidido tratar o doente com imunoglobulinas intravenosas numa dose de 2 g/kg de peso corporal repartida por 3 dias. Após apenas um ciclo de tratamento, as alterações cutâneas começaram a regredir. O escleromixedema de Arndt-Gottron é uma mucinose cutânea caracterizada por pápulas esclerodermóides e está normalmente associada a uma gamopatia monoclonal. As complicações podem incluir danos no sistema cardiovascular, no trato gastrointestinal, nos pulmões, nos rins e no sistema nervoso central.
Literatura:
- Coordenação Nacional das Doenças Raras (Kosek), www.kosekschweiz.ch, (último acesso em 03/06/2024)
- “Rare skin diseases”, www.derma.swiss/patienten/seltene-hautkrankheiten, (último acesso em 03/06/2024)
- “Keratosis palmoplantaris of the Vörner type: Clinical and formal genetic investigations”, https://archiv.ub.uni-marburg.de/diss/z2000/0196/pdf/dhb.pdf,(último acesso em 03/06/2024)
- Hoffmann JHO, Enk AH: Escleromixoedema. JDDG 2020; 18(12): 1449-1468. https://onlinelibrary.wiley.com/doi/pdf/10.1111/ddg.14319_g
- Calonje E, et al: Doenças degenerativas e metabólicas. In: Calonje E: McKees’s Pathology of the Skin. 5 ed: Elsevier, 2020: 621-622.
- Koschitzki K-T, et al. Queratose palmoplantar epidermolítica hereditária (PPK) devida a uma mutação da desmogleína-1, P077. JDDG 2024; 22, Edição S1: 1-40.
- Mann C, Butsch F, Staubach-Renz P: Terapêutica bem sucedida do escleromixedema com imunoglobulinas intravenosas, P074. JDDG 2024; 22, Edição S1: 1-40.
- Hohl D: O que é que liga as doenças de pele raras à nossa prática diária? Fórum Médico Suíço 2023; 23(12): 978-979.
- Orador E. O que é que raro e comum têm em comum? Br J Dermatol 2022; 187(3): 279-280.
- Ferreira CR: O peso das doenças raras. Am J Med Genet A 2019; 179(6): 885-892.
- Neuanfang Wakap S, et al: Estimar a prevalência pontual cumulativa de doenças raras: análise da base de dados Orphanet. Jornal Europeu de Genética Humana 2020; 28(2): 165-173.
DERMATOLOGIE PRAXIS 2024; 34(3): 43-44 (publicado em 14.6.24, antes da impressão)