Prophylactic treatment with factor concentrates is now standard in the treatment of patients with severe hemophilia A. Conventional factor VIII and factor IX concentrates have a relatively short half-life and accordingly must often be administered intravenously. For some months now, both factor VIII and factor IX products with extended half-lives have been approved in Switzerland. Various alternative therapeutic options for the treatment of hemophilia could reach market maturity in the next few years.
Although in principle all congenital coagulation disorders that lead to an increased bleeding tendency can be subsumed under the term “hemophilia”, in a narrower sense it describes two defined disorders of blood coagulation: On the one hand, the deficiency of blood coagulation factor VIII (hemophilia A) and, on the other hand, the deficiency of blood clotting factor IX (hemophilia B). Both disorders are inherited in an X-linked recessive manner. In addition to hereditary hemophilia, there are also the very rare acquired forms (autoimmune hemophilia), but these will not be discussed here.
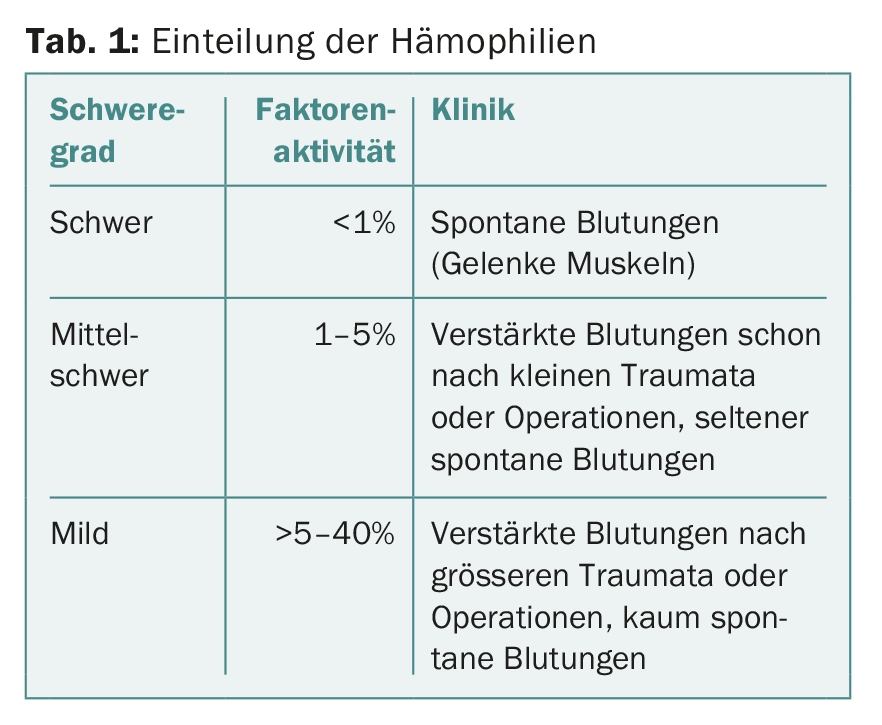
In the hereditary hemophilias, residual factor VIII or IX activity correlates relatively well with clinical presentation. This allows the classification of hemophilia A and B according to the residual activity of factor VIII and IX, respectively (Tab. 1).
Treatment of hemophilia
The invention of intravenous substitution treatment with factor concentrates from human plasma more than 50 years ago significantly reduced the mortality and morbidity of hemophilia by decreasing bleeding episodes. However, this success story was severely clouded by the transmission of viral diseases such as HIV and hepatitis C in the 1970s and 1980s. However, thanks to the implementation of better screening and virus inactivation methods and the development of recombinant factor preparations, the safety of the preparations has been massively improved, so that no virus transmissions have been reported since 1990. The main risk of treatment with factor concentrates nowadays is the occurrence of inhibitors, which are observed in about 20-30% of patients with severe hemophilia A (somewhat less frequently in hemophilia B). As the name implies, these inhibitors inhibit the supplied clotting factors and render the substitution treatment ineffective. However, in many cases it is possible to get rid of the inhibitors by means of complex immunotolerance therapies.
In patients with severe hemophilia, the so-called “prophylaxis” is standard. Factor concentrates are (usually) administered intravenously two to three times a week in such a way that spontaneous (joint) bleeding is largely prevented by increasing the plasma concentrations of the coagulation factor concerned. The frequency of injection ultimately depends on the half-life of the clotting factor involved (factor VIII approximately 12 h, factor IX approximately 18 h) [1].
The relatively frequent intravenous injections are experienced as cumbersome by many patients, and ultimately, continuous normalization of the coagulation system is still not achieved.
New factor preparations
There is, after all, some potential for improvement in the development of new therapeutic options for hemophilia: extension of the half-life (thus fewer injections and better protection), alternative forms of administration (oral or subcutaneous instead of intravenous), lower immunogenicity (less inhibitory body formation), and, in principle, better efficacy.
Certainly the most advanced are the techniques for extending the half-life. In principle, recombinant clotting factors are modified in such a way that they are degraded less rapidly. Various such “long-acting” preparations have received marketing authorization in Switzerland in recent months. The approaches used are on the one hand chemical modification of the factors (coupling to polyethylene glycols (PEGylation)), fusion with other proteins (Fc region of immunoglobulin G, recombinant albumin) or targeted protein sequence modification. For the sake of completeness, it should be mentioned here that in recent years some recombinant classical factor VIII preparations have also come onto the market which, thanks to optimized manufacturing processes (e.g. optimized glycolization), tend to have longer half-lives than their predecessors. However, these preparations are not considered “half-life extended” in the strict sense.
In the following, the methods used to achieve an extended half-life will be briefly presented.
PEGylation: In this technique, one to several 20-60 kDa PEG molecules are attached to a factor VIII or IX molecule. Different manufacturers use different concepts for this. It is essential that the PEG molecules are attached to the factor molecule in such a way that its hemostaseological effect is not impaired.
Due to the coating with PEG molecules, the factor molecules are less accessible for proteases. In addition, the PEGylation causes an increase in size as well as an improved water solubility. All this leads to a prolonged half-life of PEGylated factors [2].
Fusion protein technology: By fusing a clotting protein with another protein that naturally has a significantly longer half-life in the bloodstream, the half-life of the clotting protein itself can ultimately be increased.
Specifically, the Fc region of immunoglobulin G (IgG-Fc) or recombinant albumin are used for fusion. Both IgG-Fc and albumin have very long half-lives because they are protected from lysosomal degradation by a kind of recycling mechanism [3].
Site-specific sequence modification: In this technique, the factor VIII molecule is modified to have a significantly increased binding affinity to its carrier von Willebrand factor molecule. At around 15 hours, von Willebrand factor has a slightly longer half-life than factor VIII. Due to the strong binding of the factor VIII molecule to the von Willebrand factor molecule, the half-life of factor VIII approaches that of von Willebrand factor [4].
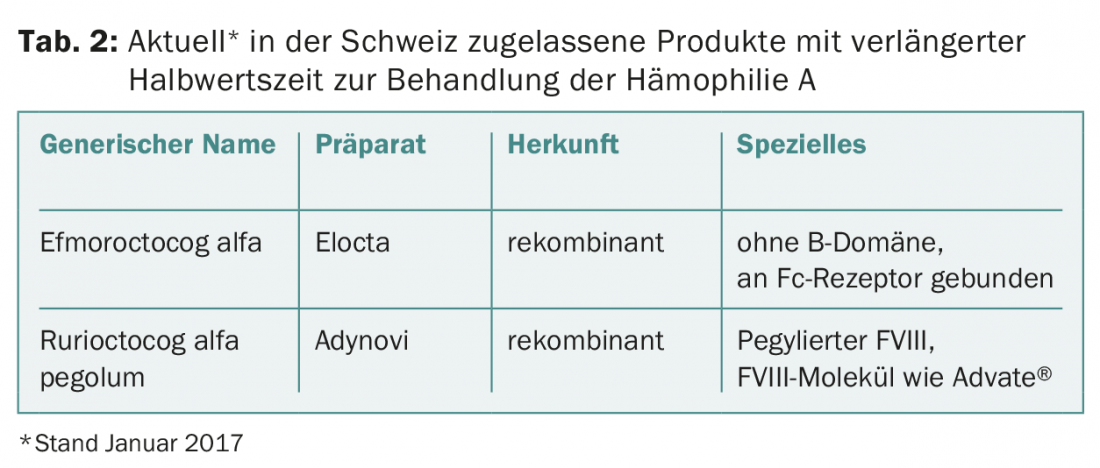
Half-life prolongation with FVIII products: The half-life of factor VIII is ultimately limited by von Willebrand factor [5]. This is because von Willebrand factor, as a carrier protein, protects factor VIII from proteolytic degradation. All the measures described above therefore make it possible at most to prolong the half-life of factor VIII (about 12 h) by about 1.5 times. Although this is not very much, it already means measurable progress clinically. For example, if a patient needs three injections per week with a conventional product, they could achieve the same valley levels with just two injections per week with a half-life-extended product. On the other hand, if the same patient maintained his injection frequency of three times per week with the new product, he could achieve significantly higher valley levels. This would be useful, for example, if he had still had breakthrough bleeding despite regular prophylaxis [6]. The half-life extended factor VIII products currently approved in Switzerland are listed in Table 2 .
Half-life extension for factor IX products: The normal blood clotting factor factor IX has a half-life of around 18 to 20 hours. Unlike factor VIII, factor IX is not bound to a carrier protein. The measures described above therefore have a much more impressive effect on factor IX than on factor VIII: both PEGylation and the fusion proteins cause an extension of the half-life by a factor of 5, i.e. to approximately 90 hours.
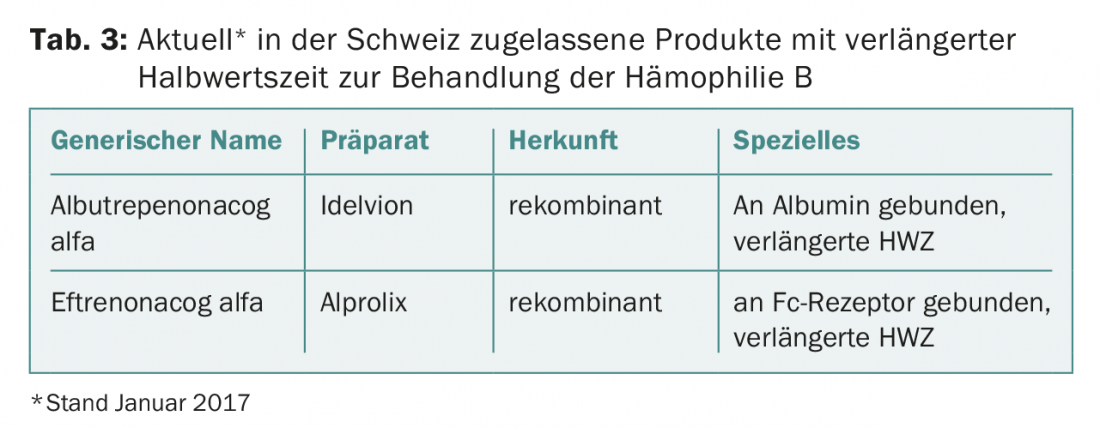
This impressive result opens up new therapeutic strategies. The half-life-extended factor IX preparations only need to be applied every one to two weeks for prophylaxis (normal factor IX preparations approx. twice a week) and higher valley levels can still be achieved [7]. The half-life extended factor IX products currently approved in Switzerland are listed in Table 3.
Alternative therapy approaches
In addition to the methods mentioned above, which ultimately all aim to optimize substitution therapy with factor concentrates, numerous alternative therapeutic approaches are now also being pursued. The aim is to achieve sufficient coagulation activation despite the existing hemophilia by targeted manipulation of the coagulation system in order to at least avoid spontaneous bleeding. Possible advantages of deviating from the classical concept of factor substitution include avoiding the problem of inhibitor development and the possibility of alternative routes of administration.
One of these alternative approaches is to block physiologic anticoagulation. Specifically, research is being conducted to specifically inhibit or downregulate tissue factor pathway inhibitor (TFPI) or antithrombin. Fitusiran, for example, is a molecule that can target antithrombin. This RNA interference molecule, which can be applied subcutaneously, specifically inhibits the formation of antithrombin. This leads to better thrombin generation in the coagulation system, resulting in better bleeding control in the patient with hemophilia [8]. The substance Fitusiran has already been successfully tested clinically (phase 1/2).
Another interesting substance that is successfully undergoing a clinical trial program to date is the bi-specific antibody emicizumab. To some extent, this antibody mimics the function of activated factor VIII by simultaneously binding to coagulation factors IX and X, resulting in increased activation of factor X and thus accelerated blood clotting. Emicizumab can be injected subcutaneously and it is also effective when inhibitors to factor VIII are present. However, the antibody does not achieve the same biological efficiency as natural coagulation factor VIII [9].
Another therapeutic approach that is still being pursued is gene therapy. In studies, it is possible to improve the endogenous activity of factor IX in patients with (severe) hemophilia B in the longer term, but normalization of factor IX activity has not yet been achieved. In hemophilia A, gene therapy is not yet technically feasible [9].
Conclusion
The many new products that have come on the market in recent months and that will come on the market in the next few years are promising. Some of the preparations mentioned definitely have the potential to revolutionize hemophilia treatment. However, it is still unclear whether, at best, the potentially high costs, especially of the alternative therapeutic approaches discussed above, could influence clinical use.
Particularly because of the numerous options available to us in developed countries for the treatment of hemophilia, we must not forget that worldwide many of these patients still do not have sufficient access to factor concentrates.
Literature:
- Ljung R: Aspects of prophylactic treatment of hemophilia. Thrombosis journal. 2016;14(Suppl 1): 30.
- Ivens IA, et al: PEGylated therapeutic proteins for haemophilia treatment: a review for haemophilia caregivers. Haemophilia: the official journal of the World Federation of Hemophilia. 2013;19(1): 11-20.
- Powell JS: Longer-acting clotting factor concentrates for hemophilia. Journal of thrombosis and haemostasis: JTH. 2015;13 Suppl 1: S167-75.
- Buyue Y, Liu T, et al: A single chain variant of factor VIII Fc fusion protein retains normal in vivo efficacy but exhibits altered in vitro activity. PloS one. 2014;9(11): e113600.
- Pipe SW, et al: Life in the shadow of a dominant partner: the FVIII-VWF association and its clinical implications for hemophilia A. Blood. 2016.
- Tiede A: Half-life extended factor VIII for the treatment of hemophilia A. Journal of thrombosis and haemostasis: JTH. 2015;13 Suppl 1: S176-9.
- Nazeef M, Sheehan JP: New developments in the management of moderate-to-severe hemophilia B. Journal of blood medicine. 2016;7: 27-38.
- Sehgal A, et al: An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nature medicine. 2015;21(5): 492-7.
- Shima M, Hanabusa H, Taki M, et al: Factor VIII-Mimetic Function of Humanized Bispecific Antibody in Hemophilia A. The New England journal of medicine. 2016;374(21): 2044-53.
InFo ONCOLOGY & HEMATOLOGY 2017; 5(1): 5-9.