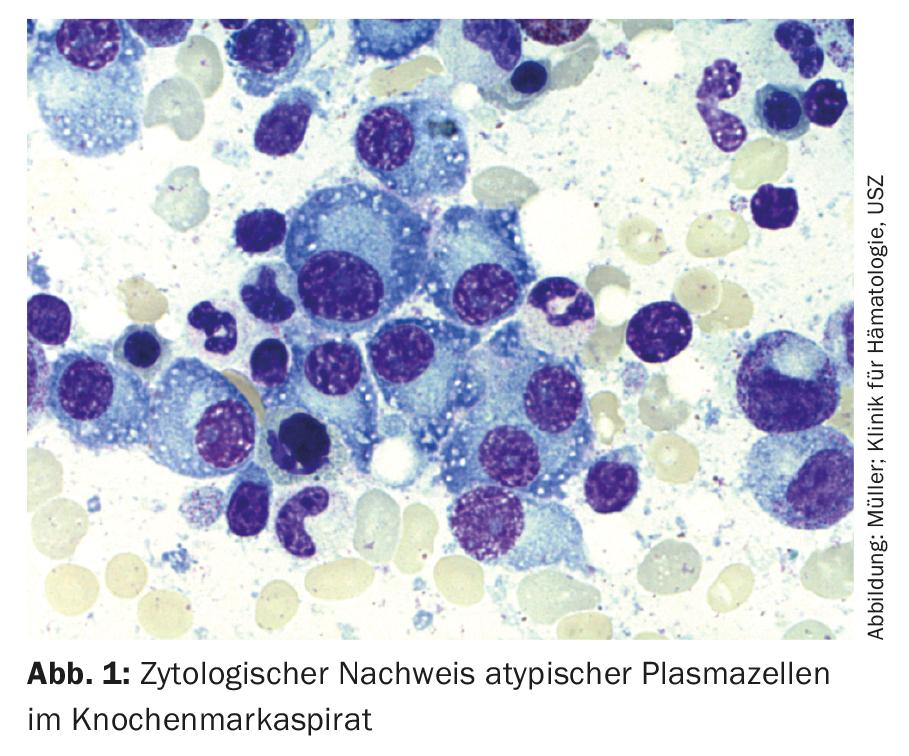
In multiple myeloma, the clinic leading to the diagnosis is derived from the end-organ damage. The diagnostic criteria were last updated in 2014. With well-established and widely available diagnostics, the disease rarely presents diagnostic difficulties today.
With an annual incidence of approximately 5-6/100,000 and a proportion of 10%, multiple myeloma is one of the most common hematologic neoplasms. With a median age of 65-70 years, older people are predominantly affected, and men are more likely to develop the disease than women, with a ratio of 1.5:1 [1].
Pathogenesis
Neoplastic transformation of a germinal center B cell in differentiation to an immunoglobulin-producing plasma cell is the disease-initiating event for a spectrum of plasma cell diseases that manifest as monoclonal gammopathy of unclear significance (MGUS), smouldering myeloma (SM), multiple myeloma (MM), or plasma cell leukemia (PCL), depending on disease activity and clinical manifestation [2].
In this context, monoclonal gammopathy of unclear significance represents a clonal precursor lesion whose incidence increases with age (occurring in approximately 3% of >70-year-olds [3]), but which progresses to multiple myeloma in only about 1% of cases per year [4,5]. The exact risk of progression here depends on the type and concentration of the paraprotein, the free light chain ratio, the clonal plasma cell percentage in the bone marrow, and immunoparesis [6,7].
In between, the stage of asymptomatic so-called smouldering myeloma can be delineated in approximately 14% of patients with an annual progression rate of 10% in the first five years after initial diagnosis, followed by 3% per year in the next five years and 1.5% in the years thereafter [8,9]. This is a clinically defined disease state between MGUS and multiple myeloma that includes a very heterogeneous patient population, including patients with MGUS-like premalignant disease progression and those with CRAB-negative aggressive multiple myeloma.
Plasma cell leukemia is the most aggressive and leukemic manifestation of plasma cell neoplasms, is comparatively rare with an incidence of approximately 4/10,000,000 [10], and may develop primarily or secondarily from preexisting multiple myeloma (1-4% of all patients) [11]. A plasma cell percentage of 20% or a concentration of 2000 plasma cells/µl of blood is required for the diagnosis in the microscopic differential blood count.
The initiating event in the oncogenesis of plasma cell dyscrasias occurs during a phase of B-cell development that is per se characterized by genetic instability due to changes in the isotype class of the immunoglobulin molecule and somatic hypermutation aimed at affinity maturation [12].
Cytogenetically, two main karyotype changes can be distinguished that are present in terms of primary mutations early in oncogenesis at the MGUS stage. Hyperdiploid karyotypes, observed in nearly two-thirds of cases, are characterized by trisomies in chromosomes with odd numbers (3,5,7,9,11,15,19) and are distinguished from the so-called non-hyperdiploid karyotype, which is often caused by translocations of the heavy chain immunoglobulin locus (IgH) with oncogenes including FGFR-3 and MMSET (t[4;14]), MAF (t[14;16]), CCND1 (t[11;14]) or characterized by unbalanced gains/losses of 1q, 1p, 6q, 8p, 13q, 16q, and 17p.
Secondary alterations include mutations in RAS proteins (K/N-RAS), activating mutations in kinases such as PI3K, AKT, BRAF, translocations involving activation of transcription factors such as MYC, and deletions or inactivation of tumor suppressor genes such as p53 and RB1 [13].
In this context, the disease is characterized by increasing clonal heterogeneity and genomic instability during its course [14], which may be enhanced by chemotherapeutic intervention (e.g., alkylants).
Once a malignant plasma cell clone is established, clinical end organ damage develops with increasing disease activity. Osteolytic bone lesions due to increased bone resorption are the result of dysregulated bone metabolism with increased osteoclast and suppressed osteoblast activity mediated by increased RANKL (“receptor activator of NF kappa B ligand”) expression, decreased expression of osteoprotegerin [15], and an osteoclast-supportive cytokine environment (increased MIP-1 alpha, IL6, IL3, etc.). Another consequence of this imbalance is the release of calcium from the bone substance with resulting serum hypercalcemia and changes in neuromuscular excitability.
Anemia, which often leads to initial diagnosis, and its workup are the result of displacement of healthy hematopoiesis by malignant plasma cells. However, this explanation falls short, because in many cases pronounced anemias are observed that cannot be explained by a proven low plasma cell infiltration. Here, alterations in the bone marrow microenvironment such as activation of the TGFβ signaling pathway appear to play a role, leading to decreased levels of hematopoietic progenitor cells in the bone marrow of myeloma patients [16].
In addition to direct plasma cell infiltration as a pathogenetic element, monoclonal paraprotein secreted by plasma cells sometimes plays a crucial role in pathogenesis. Although a hyperviscosity syndrome caused by high paraprotein concentrations is comparatively rare and is then mainly found in myeloma diseases with IgM or IgA paraprotein, which can be explained by their more complex molecular structure with occurrence as pentamer (IgM) or dimer (IgA), the end organ damage caused by toxic light chain amyloid in the pathogenesis of AL amyloidosis, however, already occurs at low paraprotein concentrations. Especially in light chain myeloma, glomerular filterable free light chains lead to tubular damage and obstruction (so-called cast nephropathy) due to their smaller molecular size compared to the complete immunoglobulin molecule.
Clinic
The clinical symptoms that ultimately lead to physician consultation and diagnosis are derived from the myeloma-related end-organ damage described in their pathogenesis. Frequently, bone pain due to myeloma-related osteolysis, a pathologic osteolytic-related fracture, impaired performance with anemia, or a tendency to infection lead to medical presentation. The nephrotoxic effect of the light chains can lead to renal insufficiency up to renal failure with corresponding uremic symptoms. Less commonly, multiple myeloma manifests with cardiac arrhythmias, somnolence, or other hypercalcemia-related symptoms. The toxic effect of light chain amyloid in AL amyloidosis can lead to a very diverse clinical symptomatology. Cardiac insufficiency due to cardiac deposition is often found here, along with renal insufficiency, polyneuropathy and others.
Diagnostics
The diagnostic criteria for multiple myeloma were last updated in 2014 by the International Myeloma Working Group (IMWG) as part of a consensus update [17].
Established CRAB criteria (hypercalcemia, renal failure, anemia, osteolysis) were supplemented by the so-called SLiM criteria (Tab. 1) . The background for the extension of the diagnostic criteria is the observation that in the collective of patients with smouldering myeloma not previously requiring treatment, certain disease parameters are associated with a high probability of progression (>80% within two years) are associated with multiple myeloma requiring treatment, and this patient group benefits from earlier therapeutic intervention [17,18].

Thus, for the diagnosis of multiple myeloma, the detection of ≥10% clonal plasma cells in bone marrow biopsy or aspirate (Fig.1) Or a biopsy-documented osseous or extramedullary plasmacytoma required, in conjunction with evidence of one or more end-organ lesions or biomarkers of malignancy (Tab.1). In this case, the clonality of the plasma cells is demonstrated by flow cytometric detection of cytoplasmic light chain restriction. (Fig. 2) or by immunohistochemical staining of the light chains on a representative bone marrow punch biopsy.
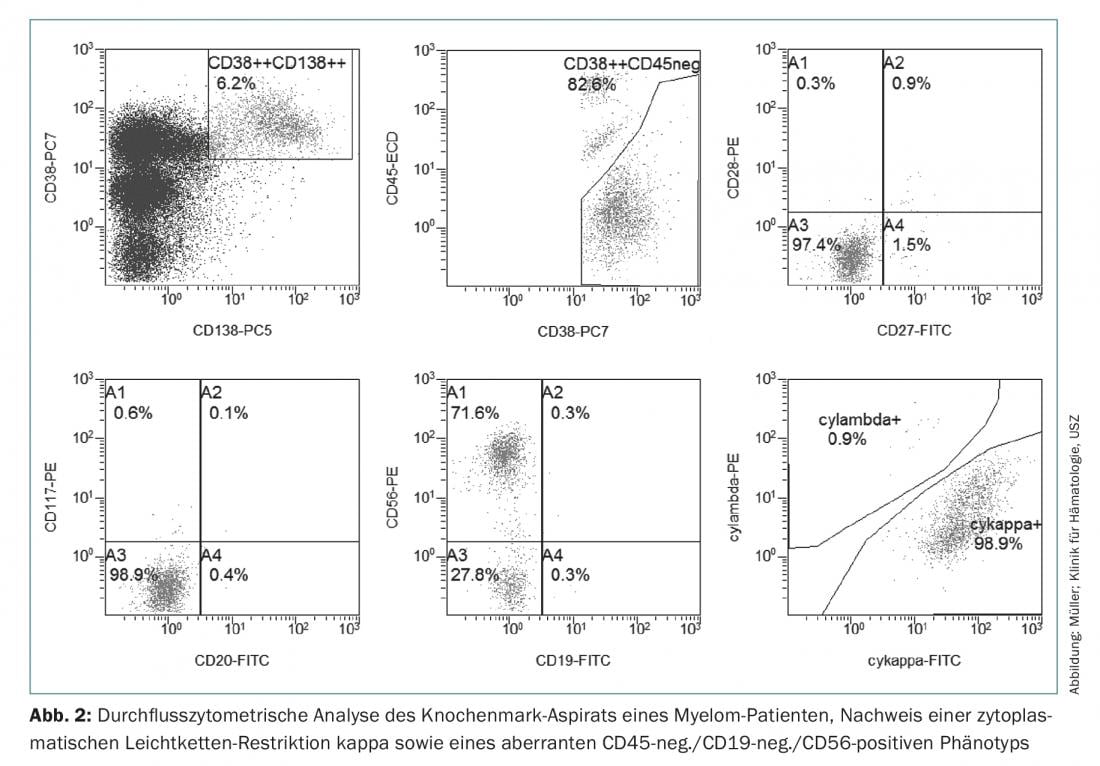
Immunophenotypically, myeloma cells can be distinguished from healthy CD38++, CD138+, CD19+, CD45+ and CD56-negative plasma cells by CD45 negativity or reduced expression, a loss of CD19 and expression of aberrant markers such as CD56, CD117 or CD28, which can be used in initial, but especially in MRD diagnostics and in some cases also has prognostic significance [19].
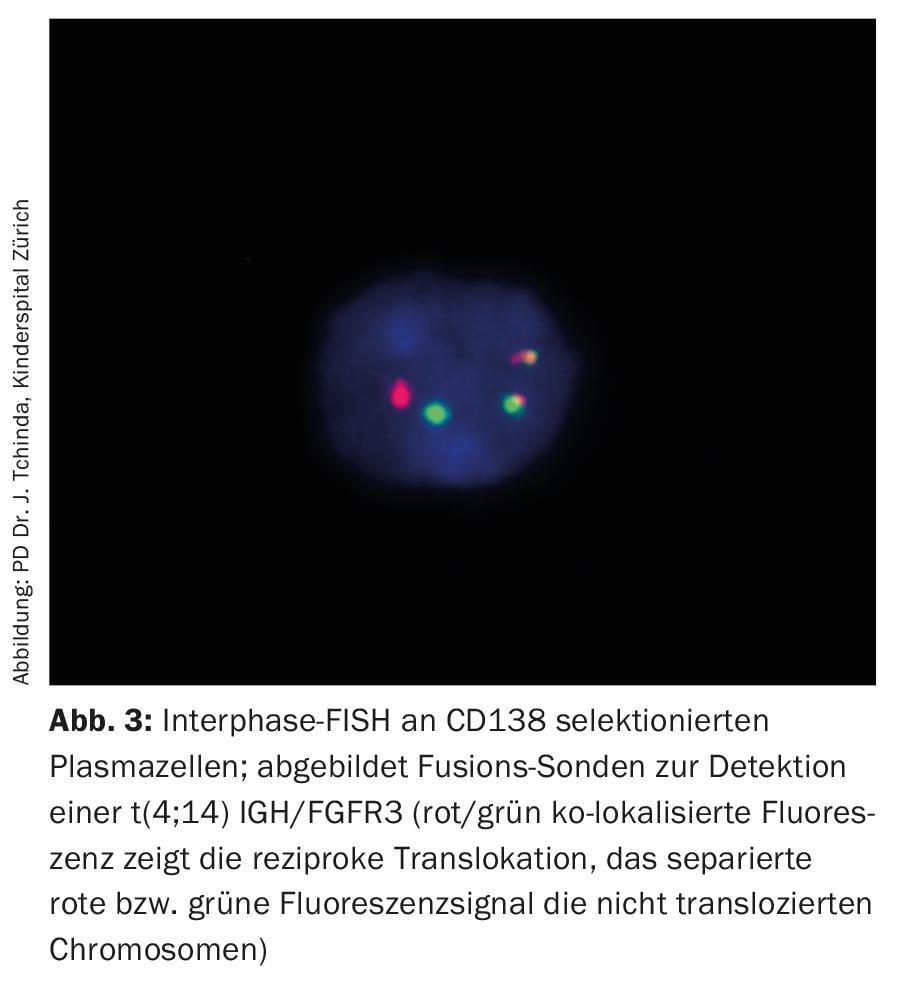
While conventional analysis of the karyotype of arrested metaphases in multiple myeloma usually remains uninformative due to the low proliferative index and difficult cell culture conditions, fluorescence in situ hybridization of interphase nuclei with fluorescent probes is part of the standard workup and, together with established surrogate parameters of disease activity such as β2-microglobulin, albumin, and lactate dehydrogenase (LDH), allows a priori risk stratification (Fig. 3, Tab. 3) [20].
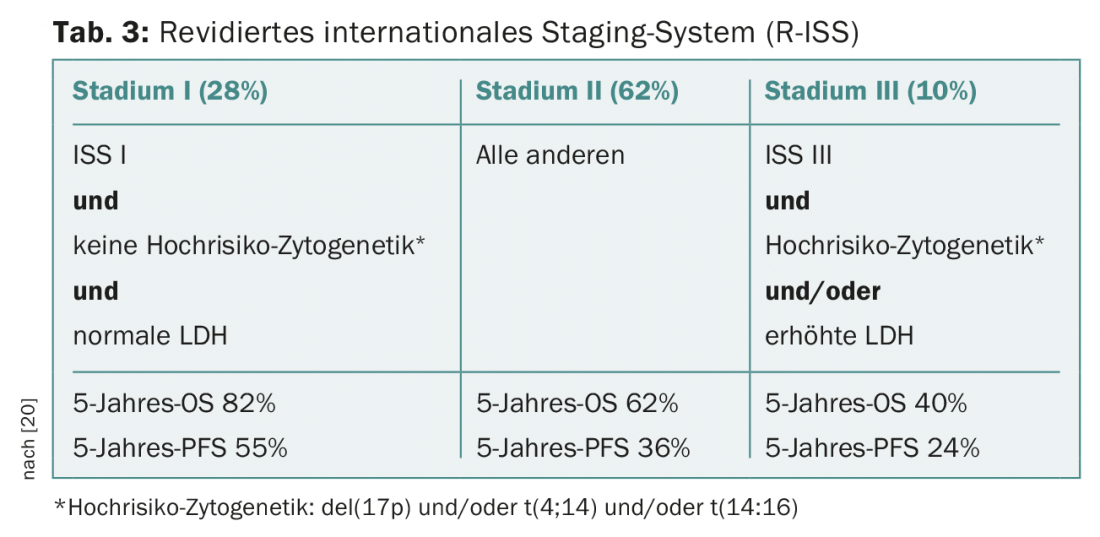
Table 2 provides an overview of the laboratory diagnostics to be performed at initial diagnosis as well as the necessary imaging by whole-body low-dose CT and optionally MRI and PET-CT.
With well-established and widely available diagnostics, multiple myeloma rarely presents diagnostic difficulties in modern times. Rather, the future challenges lie in an increasingly determined genetic subclassification of the disease entity with the goal of improved risk stratification and the establishment of predictive markers for treatment response.
Furthermore, in the context of increasingly effective therapeutic options with improved response and deeper remissions, the detection of minimal residual disease (MRD) by flow cytometry and next generation sequencing is playing an increasingly important role in response and remission assessment. While the prognostic significance of MRD for progression-free survival (PFS) and overall survival (OS) has already been demonstrated [21], clinical MRD-based treatment decisions are not yet established but are the subject of current clinical trials.
Take-Home Messages
- The clinic leading to physician consultation and diagnosis is derived from the myeloma-related end-organ damage.
- The diagnostic criteria for multiple myeloma were last updated in 2014 as part of a consensus update.
- With well-established and widely available diagnostics, multiple myeloma rarely presents diagnostic difficulties today.
- Rather, the future challenge lies in an increasingly determined genetic subclassification of the disease entity with the goal of improved risk stratification and the establishment of predictive markers for treatment response.
- The prognostic significance of minimal residual disease (MRD) for progression-free survival (PFS) and overall survival (OS) has been demonstrated, and clinical MRD-based treatment decisions are the subject of current clinical trials.
Literature:
- Rodriguez-Abreu D, Bordoni A, Zucca E: Epidemiology of hematological malignancies. Annals of Oncology 2007; 18(Suppl 1): i3-i8.
- Bakkus MH, et al: Evidence that multiple myeloma Ig heavy chain VDJ genes contain somatic mutations but show no intraclonal variation. Blood 1992; 80: 2326-2335.
- Kyle RA, et al: Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med 2006; 354: 1362-1369.
- Landgren O, et al: Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood 2009; 113: 5412-5417.
- Weiss BM, et al: A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 2009; 113: 5418-5422.
- Baldini L, et al: Role of different hematologic variables in defining the risk of malignant transformation in monoclonal gammopathy. Blood 1996; 87: 912-918.
- Turesson I, et al: Monoclonal gammopathy of undetermined significance and risk of lymphoid and myeloid malignancies: 728 cases followed up to 30 years in Sweden. Blood 2014; 123: 338-345.
- Kristinsson SY, Holmberg E, Blimark C: Treatment for high-risk smoldering myeloma. N Engl J Med 2013; 369: 1762-1763.
- Kyle RA, et al: Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med 2007; 356: 2582-2590.
- Sant M, et al: Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood 2010; 116: 3724-3734.
- Tiedemann RE, et al: Genetic aberrations and survival in plasma cell leukemia. Leukemia 2008; 22: 1044-1052.
- Seifert M, Scholtysik R, Küppers R: Origin and pathogenesis of B cell lymphomas. Methods Mol Biol 2013; 971: 1-25.
- Kuehl WM, Bergsagel PL: Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer 2002; 2: 175-187.
- Bolli N, et al: Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun 2014; 5: 2997.
- Roodman GD: Mechanisms of bone metastasis. N Engl J Med 2004; 350: 1655-1664.
- Bruns I, et al: Multiple myeloma-related deregulation of bone marrow-derived CD34(+) hematopoietic stem and progenitor cells. Blood 2012; 120: 2620-2630.
- Rajkumar SV, et al: International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15: e538-48.
- Mateos MV, et al: Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med 2013; 369: 438-447.
- Mateo G, et al: Prognostic Value of Immunophenotyping in Multiple Myeloma: A Study by the PETHEMA/GEM Cooperative Study Groups on Patients Uniformly Treated With High-Dose Therapy. Journal of Clinical Oncology 2008; 26: 2737-2744.
- Palumbo A, et al: Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol 2015 Sep 10; 33(26): 2863-2869.
- Paiva B, van Dongen JJM, Orfao A: New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood 2015; 125: 3059-3068.
- Moreau P, et al: Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up? Ann Oncol 2017 Jul 1; 28(suppl_4): iv52-iv61.
InFo ONCOLOGY & HEMATOLOGY 2017; 5(5): 7-10.