Immune-mediated neuropathies vary in clinical presentation, course, and pathogenesis. In recent years, autoantibodies against nodal and paranodal proteins of the Ranvier lacing ring have been detected. The autoantibodies can cause axonal damage and lead to demyelination.
Immune-mediated neuropathies vary in clinical presentation, course, and pathogenesis and account for approximately 9% of all polyneuropathies [1]. In recent years, autoantibodies against nodal and paranodal proteins of the Ranvier lacing ring have been detected. Autoantibodies against neurofascin (NF), contactin 1 (CNTN1), or contactin-associated protein 1 (CASPR1) were detected in 10% of patients with chronic inflammatory demyelinating polyneuropathy (CIDP) [2]. The autoantibodies can cause axonal damage and lead to demyelination [4]. Seropositive patients with CIDP differ from seronegative patients in clinical phenotype, electrophysiologic findings, and response to standard therapy [2].
Detection of specific nodal and paranodal autoantibodies may help identify effective therapies. In addition to CIDP, disease-associated autoantibodies are also known in other immune-mediated neuropathies, such as Guillain-Barré syndrome (GBS) with the subtypes acute inflammatory demyelinating polyneuropathy (AIDP), acute motor axonal neuropathy (AMAN), and acute motor and sensory axonal neuropathy (AMSAN), Miller-Fisher syndrome (MFS), multifocal motor neuropathy (MMN), anti-MAG MGUS neuropathy (paraproteinemic neuropathy), sensitive neuronopathy in systemic immune diseases, and paraneoplastic polyneuropathies.
Pathophysiology and electrophysiological alterations of nodo- and paranodopathies.
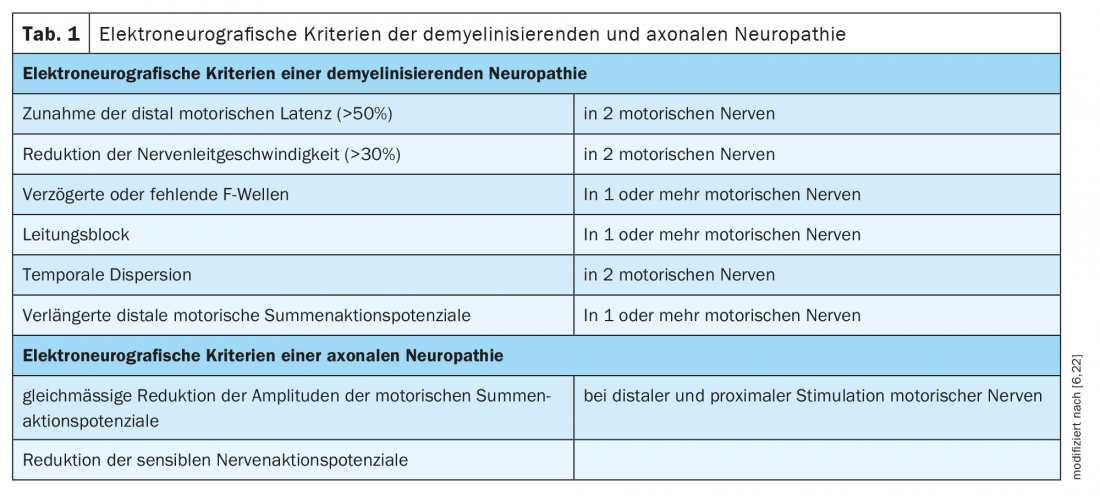
Polyneuropathies are traditionally divided into demyelinating and axonal neuropathies according to criteria in electrophysiology (Table 1). The underlying pathology is expected to be at the myelin/swan cell or axon accordingly. The proteins of the nodal or paranodal region of Ranvier’s lacing ring are junctions between the myelin/microvilli of the Schwan cell and the axon (Fig. 1) . In neuropathies due to autoantibodies against nodal and paranodal proteins, a clear assignment to a demyelinating or axonal pattern is not possible due to the underlying pathomechanism. The term nodo- and paranodopathies was proposed by Uncini, Susuki, and Yuki in 2013 [8,9].
Features of nodo- and paranodopathy are
- Antibodies to nodal and paranodal proteins have different etiologies, but all result in dysfunction in the nodal region of Ranvier’s lacing ring.
- Pathological continuum from transient conduction block to axonal degeneration.
- Conduction block due to detachment of myelin from the axon, widening of the nodal region, or dysfunction/disruption of sodium channels (high density in the nodal region for saltatory stimulus conduction) with segmental abnormal polarization of the axolem.
- Conduction block may be rapidly reversible without appearance of marked temporal dispersion; conduction block may also be preserved.
- Axonal degeneration is dependent on the specific disease and severity, possibly followed by conduction block.
- Diagnosis can be made by repeated electrophysiological studies:
- a. in acute disease, in the presence of a rapidly reversible conduction block or reduction in nerve conduction velocity without marked temporal dispersion or progression of conduction block to axonal degeneration;
- b. in chronic disease with persistent conduction block and additional signs of axonal degeneration.
The use of the term nodo- and paranodopathies puts the focus on the localization of the primary nerve damage and helps to better classify the segmental demyelinating neuropathy, the paradox of reversible axonal damage and the good prognosis, despite axonal abnormalities in electrophysiology [9].
Autoantibodies in chronic inflammatory demyelinating polyneuropathy (CIDP).
Localization: In the paranodal region, the complex of contactin 1 and CASPR1 on the axon and neurofascin 155 on the glial cell of the myelin sheath consolidates the contact between the myelin processes and the axon (Fig. 1) [10].
Anti Contactin 1 (CNTN1) IgG
- Symptoms: 20 – 49% show a subacute onset of disease. A GBS-like aggressive symptom onset is known with subacute onset paresis (Casuistics). A predominantly motor neuropathy is typical and sensory ataxia is frequently observed. The occurrence of tremor is more common than in seronegative CIDP but less common than in anti NF155-positive CIPD. The age of onset is around 50-60 years. Year of life. Overall, a higher age of anti-CNTN1-positive CIDP patients at disease onset than seronegative CIDP patients has been described [2].
- Findings: Electroneurographic studies reveal axonal changes early in the course of the disease (Table 1) .
- Therapy implications: Patients respond inadequately to intravenous immunoglobulins (IVIg). Corticosteroids have a good effect. If there is an inadequate response to standard therapy, B-cell-depleting therapy with rituximab is effective [14]. It should be noted that the shortest possible duration of disease is important to minimize axonal damage [8]. Under effective immunotherapy, serum titers of anti CNTN1 decrease [11].
Anti Contactin Associated Protein 1 (CASPR1) IgG
- Symptoms: Marked neuropathic pain has been described in anti CASPR1-seropositive patients. Symptoms usually start subacute with severe onset and are initially motor accentuated. Anti CASPR1 has been detected in patients with CIDP and GBS. The onset of the disease is reported in a case report around the age of 30.
- Findings: Reversible conduction blocks can be detected in electrophysiological studies [11].
- Therapy implications: Lack of response to IVIg and methylprednisolone has been described. Rituximab has shown very good efficacy. Associated neuropathic pain improves with effective therapy [2].
Anti Neurofascin 155 (NF155) IgG
- Symptoms: Typically, a high proportion of NF155-positive CIDP patients have distally emphasized weakness. Other symptoms may include high-amplitude and low-frequency tremor and sensory ataxia with cerebellar signs. The association with HLA-DRB1*15 has been described [12]. The age of onset is much younger than in seronegative CIPD with an age around 20-30 years at onset.
- Findings: Electrophysiological examination revealed a demyelinating pattern with marked prolongation of distal latencies and F-wave latencies (Table 1). A highly elevated protein was detected in the CSF [13].
- Therapy implications: Response to IVIg is poor. Partial improvement was described on corticosteroids. Good therapeutic success can be expected with rituximab and plasmapheresis [14]. Under effective immunotherapy, serum titers of anti NF155 decrease, and a correlation with clinical improvement could be shown [11].
Anti Neurofascin 140/186 (NF 140/186) IgG
- Symptoms: Patients present with -a symmetric sensorimotor polyradi-kulopathy with a severe course. In addition, sensory ataxia and cranial nerve involvement may occur. Some patients had concomitant autoimmune disease. The age of onset is around 50-60 years. year of life described [2].
- Findings: Electroneurographs revealed demyelinating findings with conduction blocks (in 3/5 patients) and axonal features (in 2/5 patients) [15].
- Therapy implications: On IVIg and corticosteroids, symptoms partially improved. A potentially good response has been described to rituximab [14].
Autoantibodies in acute inflammatory polyneuropathies – Guillain-Barré syndrome (GBS) with subtypes AIDP, AMAN and AMSAN.
Localization: the ion channels of Ranvier’s lacing ring are stabilized by gangliosides, glycosphingolipids contained in the cell membrane of the nodal and paranodal regions [16]. The various subtypes of Guillain-Barré syndrome are each associated with autoantibodies to different gangliosides and differ in electrophysiologic findings.
Acute inflammatory demyelinating poly-neuropathy (AIDP) is the most common subtype of GBS in Europe and North America. In most cases, no specific autoantibodies can be detected.
- Symptoms: The disease is often preceded by a gastrointestinal or respiratory infection. Initially, hypesthesias, paresthesias, paresis, and pain occur in the extremities. The pareses are bilateral, symmetrical, and progressive. Motor symptoms develop within 12 hours to 28 days to tetraparesis with respiratory muscle involvement. In addition, cranial nerve deficits and autonomic dysfunction may occur [4].
- Findings: This demyelinating subtype of acute inflammatory polyneuropathy is characterized by marked reduction in nerve conduction velocity, increase in distal motor latencies, conduction blocks, abnormal temporal dispersion, prolonged F-wave latencies, or absence of F-waves [17].
Anti LM1 (sialosylneolactotetraosylceramide)
Autoantibodies against LM1 (sialosylneolactotetraosylceramide) cause acute inflammatory demyelinating polyneuropathy (AIDP) when monospecific. Cases have been described in which anti LM1 IgG antibodies cause a cross-reaction with gangliosides such as GM1, GalNAc-GD1a, GD1b and GQ1b, so that in that case the diagnosis of AMAN or AMSAN can be made.
Anti GalC (Galactocerebrosides)
Autoantibodies to galactocerebrosides (GalC) have been detected in children with M. pneumoniae-associated severe acute inflammatory demyelinating polyneuropathy (AIDP) [18].
Anti neurofascin 186, anti gliomedin, anti NrCAM, anti CNTN1, anti neurofascin 155, anti CASPR1 and anti CASPR2.
In studies, the nodal autoantibodies anti neurofascin 186, anti gliomedin, and anti NrCAM, and the paranodal autoantibodies anti CNTN1, anti neurofascin 155, and anti CASPR1 (see above under the CIDP section) have been detected in adult patients with GBS. Juxtaparanodal autoantibodies to CASPR2 have been described in 2 cases of GBS in children.
Acute motor axonal neuropathy (AMAN) is most common in Asia [19].
- Symptoms: Corresponding to AIDP (see above) without sensory disturbances.
- Findings: This axonal subtype of acute inflammatory polyneuropathy is characterized by marked reduction in motor cumulative action potential (CMAP) amplitudes and so-called “reversible failure of conduction,” i.e., reduced CMAP amplitude and conduction blocks may suddenly recover on repeated electroneurographic examination without evidence of temporal dispersion as a sign of remyelination [16].
Anti-GM1 IgG, Anti-GM2, Anti-GD1b IgG, Anti-GT1b, Anti- GM3, Anti-GD1a IgG and Anti-GalNac-GD1a
The ganglioside antibodies anti-GM1 IgG, anti-GM2, anti-GD1b, anti-GT1b, anti-GM3, anti-GD1a IgG, and anti-GalNac-GD1a can be detected in approximately 80% of patients with AMAN. These autoantibodies may occur singly or in combination.
Acute motor and sensory axonal neuropathy (AMSAN).
- Symptoms: see AIDP
- Findings: Same criteria as AMAN plus reduction in amplitude of the sensory nerve action potential (Unicini et al. 2018).
anti-GM1 IgG, anti-GM1b, anti-GD1a IgG
The ganglioside antibodies anti-GM1, anti-GM1b, anti-GD1a are detectable in acute motor and sensory axonal neuropathy (ASMAN) [19].
Autoantibodies in Miller Fisher Syndrome (MFS)
Localization: Ganglioside GQ1b is mainly found in the paranodal myelin of cranial nerves supplying the -eye muscles [19].
Anti GQ1b IgG
- Symptoms: Acute ophthalmoplegia, acute ataxic neuropathy, and areflexia are the main symptoms of Miller-Fisher syndrome. In Bickerstaff brainstem encephalitis, in addition to ophthalmoplegia and ataxia, there is impaired consciousness and usually hyperreflexia.
GQ1b autoantibodies can be detected in 90% of patients with Miller-Fisher syndrome (MFS). Miller-Fisher syndrome and Bickerstaff brainstem encephalitis are associated with anti GQ1b, so Shahrizaila and Yuki group the disorders together as anti-GQ1b antibody syndrome [20].
Autoantibodies in multifocal motor neuropathy (MMN).
Localization: Ganglioside GM1 is mainly localized in the nodal region of Ranvier’s lacing ring. Autoantibodies bind in the nodal region and activate complement, impairing sodium channel clustering.
Anti GM1 IgM and Anti GM1-Galactocerebroside Complex
- Symptoms: Slowly increasing, mostly asymmetric paresis of the extremities is characteristic, often starting in the upper extremities. Sensitive deficits are not found. In addition, holding tremor, fasciculations, and spasms may occur.
- Findings: Anti-GM1 IgM antibodies are detectable in 50% of all patients with MMN [21]. Conduction blocks can be detected in electrophysiological studies. Complement activation may result in axonal damage.
- Therapy implications: Good response to IVIg. Detection of autoantibodies can support the diagnosis of MMN when not all criteria are met and help initiate effective therapy with IVIg. Clinically, there is 2nd motor neuron disease, making MMN an important differential diagnosis of amyotrophic lateral sclerosis [11].
Autoantibodies in MGUS-Polyneuropathy (MGUS-P).
Localization: Myelin-associated glucoprotein (MAG) is localized in the myelin of the paranodal region.
Anti MAG (myelin-associated glucoprotein) IgM
- Symptoms: A slowly progressive predominantly sensitive ataxic distal polyneuropathy is characteristic. The upper distal extremities are commonly involved [11]. Most patients are younger than 50 years old.
- Findings: A demyelinating pattern is detectable on electrophysiologic examination. Immunofixation reveals IgM monoclonal gammopathy. Anti MAG IgM antibodies are positive in 50% of patients with MGUS-P. Antibody titer levels do not appear to correlate with disease severity and response to therapy. Detection of anti MAG autoantibodies is only required for diagnosis.
- Therapy implications: A few studies have shown response to plasmapheresis, cyclophosphamide, IVIg, and rituximab. There is evidence that early B-cell depletion with rituximab may influence progression [11].
Autoantibodies associated with systemic immune diseases
SSA (Ro) and SSB (La) antibodies, antineuronal antibodies, FGFR3 (fibroblast growth factor receptor 3) antibodies
- Symptoms: In (younger) patients with progressive, acute or subacute course of sensory neuropathy/neuronopathy, search for Sjögren’s syndrome or screening for autoimmune diseases is useful [22].
- Findings: There is a mild increase in protein with normal cell count in the CSF. Electrophysiologically, abnormalities in sensitive nerves that are not length-dependent are detectable. Extensive loss or low amplitude of sensory nerve action potentials is typical as well as an asymmetric clinical distribution (“patchy”). Occasionally, the sensitive nerves in the arms may be affected more than those in the legs [23].
- Therapy implications: The underlying disease must be treated.

In what clinical presentation should autoantibody-mediated genesis be considered?
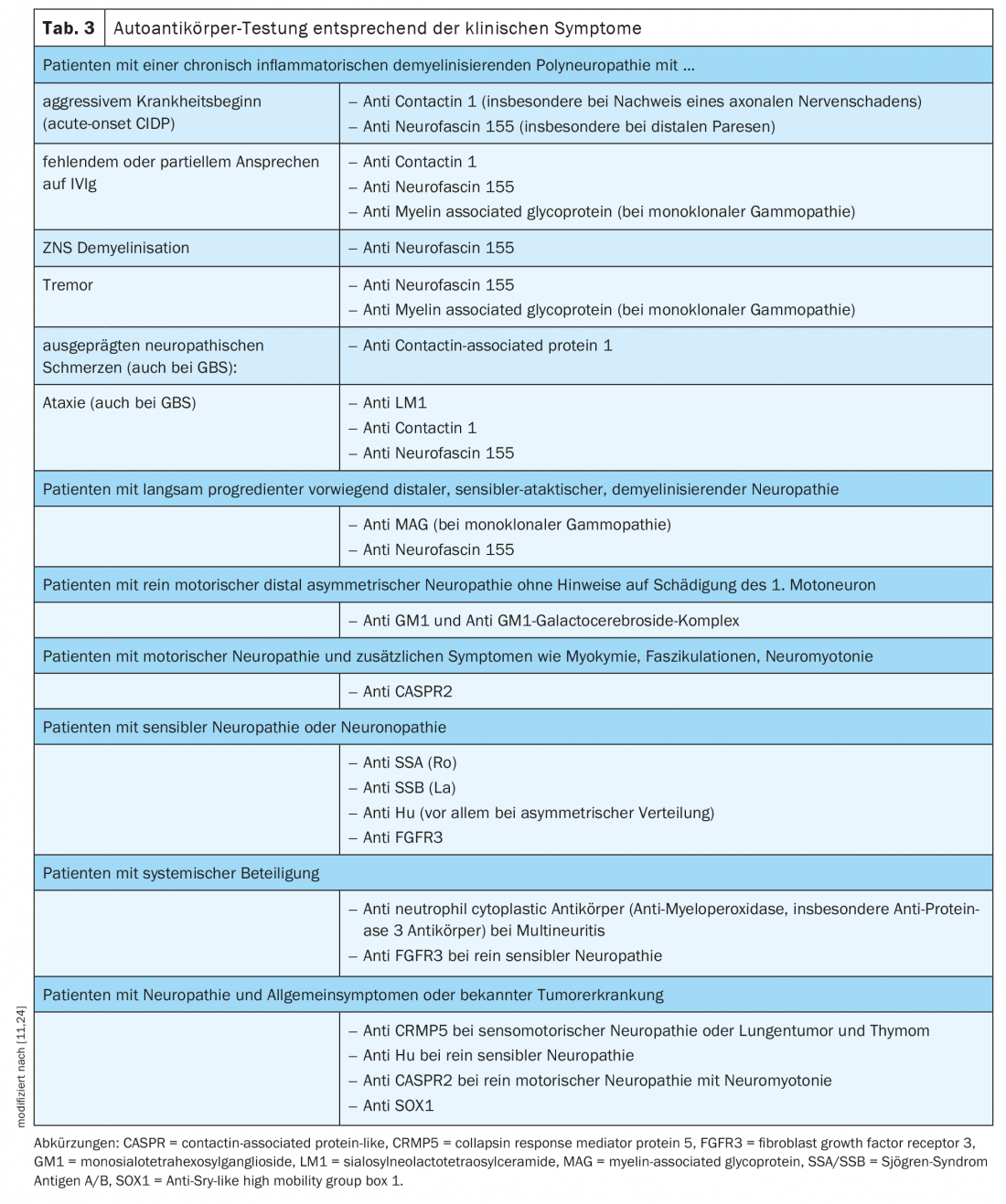
Testing for nodal and paranodal autoantibodies should be performed in patients with an acute, subacute, or chronic course of acquired demyelinating polyneuropathy associated with additional symptoms such as tremor, distal involvement, or poor response to IVIg [11] (Table 3).
Why does standard therapy often have an inadequate effect in seropositive chronic inflammatory demyelinating polyneuropathy?
Studies have shown that nodal and paranodal autoantibodies belong to different IgG subclasses (Table 4) . IgG2 and IgG3 have been found in monophasic anti CNTN1 and CASPR1 seropositive GBS, IgG4 so far only in chronic courses. From Appeltshauser et al. it has been described that in the course of disease a class change from IgG3 to IgG4 can occur in anti CNTN1 and anti CASPR1 seropositive neuropathy [25]. Currently, the IgG subclasses of nodal and paranodal autoantibodies are determined only in research laboratories.

IgG3-mediated neuropathies respond well to standard IVIg therapy. IVIg is effective in inflammatory polyneuropathies, particularly through complement inhibition and antibody neutralization. Patients with IgG2 and IgG4 antibodies either show no improvement with IVIg infusions or the response decreases during the course of the disease. The poor or absent response to IVIg in IgG4-mediated disease is attributed to the low capacity to bind FcγIIb receptors and the lack of complement activation [14]. In IgG4-mediated neurologic and non-neurologic autoimmune diseases such as anti-muscle positive myasthenia gravis, autoimmune pancreatitis, and sclerosing cholangitis, the efficacy of rituximab has been demonstrated in numerous studies. The B-cell-depleting effect of rituximab is critical in IgG4-mediated disease. A partial response of patients with nodal and paranodal IgG4 antibodies to corticosteroids has been reported [14]. In autoantibody-positive neuropathies, rapid and effective therapy is needed before irreversible damage occurs. In the absence of response to standard therapy (IVIg & steroids) for chronic inflammatory demyelinating polyneuropathy, therapy with rituximab should be considered.
Take-Home Messages
- Within the immune-mediated neuropathies, the nodo-/paranodopathies should be considered for differential diagnosis, as effective therapeutic options exist.
- Laboratory chemistry can detect autoantibodies against nodal and paranodal proteins of Ranvier’s lacing ring in nodo- and paranodopathies.
- Electroneurographic features are defined for nodo- and paranodopathies.
- The basis of the diagnosis of autoantibody-associated neuropathies are electroneurographic examinations (demyelinating or axonal pattern, features of nodo- and paranodopathies) and laboratory and CSF diagnostics (exclusion of relevant differential diagnoses, detection of cytalbuminous dissociation in CSF); in addition, nerve sonography (pattern of nerve swelling) is helpful.
- Test nodal and paranodal autoantibodies from serum (no intrathecal production and low titers in CSF).
- In the absence of improvement with first-line therapy (IVIg, steroids) and in patients with an acute or subacute course of acquired demyelinating polyneuropathy with additional symptoms such as tremor, ataxia, and distal involvement (see Table 2: Red Flags in CIDP), consider determination of autoantibodies and therapy with rituximab if appropriate.
- Selection of autoantibodies based on clinical symptoms (Table 3).
Literature:
- Visser NA: Incidence of polyneuropathy in Utrecht, the Netherlands. Neurology 2015, Jan 20; 84(3): 259-264.
- Vural A, Doppler K: Autoantibodies against the node of Ranvier in seropositive chronic inflammatory demyelinating polyneuropathy: diagnostic, pathogenic, and therapeutic relevance. Front. Immunol. 2018, May 14; 9: 1029
- Stathopoulos P: Autoimmune antigenic targets at the node of Ranvier in demyelinating disorders. Nat Rev Neurol. 2015 Mar; 11(3): 143-156.
- Kieseier BC, Mathey EK, Sommer C: Immune-mediated neuropathies. Nat Rev Dis Primers 2018 4, 31.
- Svennerholm L: Ganglioside designation. Adv Exp Med Biol. 1980; 125: 11
- Grether NB, et al: Diagnostics of immune-mediated polyneuropathies. DGNeurology 2020; 3 (2): 147-158.
- Uncini A, Kuwabara S: Electrodiagnostic criteria for Guillain-Barre syndrome: a critical revision and the need for an update. Clin Neurophysiol, 2012, 123(8), 1487-1495.
- Uncini A, Kuwabara S: Nodopathies of the peripheral nerve: an emerging concept. J Neurol Neurosurg Psychiatry, 2015, 86(11), 1186-1195.
- Uncini A, Susuki K, Yuki N: Nodo-paranodopathy: beyond the demyelinating and axonal classification in anti-ganglioside antibody-mediated neuropathies. Clin Neurophysiol, 2013, 124(10), 1928-1934.
- Poliak S, Peles E: The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci, 2003, 4(12), 968-980.
- Querol L: Autoantibodies in chronic inflammatory neuropathies: diagnostic and therapeutic implications, Nat Rev Neurol. Neurology, 2017, Sep; 13(9): 533-547.
- Martinez-Martinez L: Anti-NF155 chronic inflammatory demyelinating polyradiculoneuropathy strongly associates to HLA-DRB15. J Neuroinflammation 2017; 14: 224.
- Kadoya M: IgG4 anti-neurofascin155 antibodies in chronic inflammatory demyelinating polyradiculoneuropathy: Clinical significance and diagnostic utility of a conventional assay. Journal of Neuroimmunology 2016 Dec 15; 301: 16-22.
- Bunschoten C: Progress in diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy, Lancet Neurology 2019; 18: 784-94.
- Delmont E, Manso C, Querol L, et al: Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain. 2017 Jul 1;140(7): 1851-1858.
- Susuki K: Gangliosides contribute to stability of paranodal junctions and ion channel clusters in myelinated nerve fibers. Glia, 2007a, 55(7): 746-757.
- Uncini A, Kuwabara S: The electrodiagnosis of Guillain-Barré syndrome subtypes: Where do we stand? Clinical Neurophysiology. 2018 Dec; 129(12): 2586-2593.
- Meyer Sauteur PM: Mycoplasma pneumoniae triggering the Guillain-Barré syndrome: A case-control study, Ann Neurol 2016 Oct; 80(4): 566-580.
- Pei S: Axonal variants of Guillain-Barré-syndrome: an update. Springer Nature 2020 March.
- Shahrizaila N, Yuki N: Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013, 84: 576-583.
- Van Asseldonk JT: Multifocal motor neuropathy. Lancet Neurol, 2005, 4(5): 309-319.
- Heuss D: Diagnostics in polyneuropathies, S1 guideline, 2019, in: German Society of Neurology (ed.), Guidelines for Diagnostics and Therapy in Neurology. Online: www.dgn.org/leitlinien. (accessed 04/01/2021).
- Sghirlanzoni A: Sensory neuron diseases. The Lancet Neurology. 2005; 4(6): 349-361
- Sun X: Anti-SOX1 Antibodies in Paraneoplastic Neurological Syndrome. J Clin Neurol. 2020 Oct;16(4):530-546.
- Appeltshauser L: Antiparanodal antibodies and IgG subclasses in acute autoimmune neuropathy. Neurol Neuroimmunol Neuroinflamm. 2020 Jul 24; 7(5): e 817.
- Ilyas A: Immunoglobulin G subclass distribution of autoantibodies to gangliosides in patients with Giullain-Barre syndrome. Res Commun Mol Pathol Pharmacol. 2001 Jul; 109(1-2): 115-123.
- Lardone RD: Neurological disorders-associated anti-glycosphingolipid IgG antibodies display differentially restricted IgG subclass distribution. Sci Rep. 2020 Aug 4;10(1): 13074.
InFo NEUROLOGY & PSYCHIATRY 2021; 19(3): 19-25.