Las pruebas genéticas permiten ahora realizar diagnósticos que antes no podían hacerse. Sin embargo, depende del método, qué significado se esconde tras los resultados.
Las enfermedades cardiovasculares hereditarias, como el síndrome de Marfan (SMF), pertenecen a las enfermedades monogénicas y se consideran raras con una frecuencia (prevalencia) de <1:2000. Sin embargo, dado que existen muchas enfermedades cardiovasculares hereditarias diferentes, el número total de personas afectadas es relativamente grande y requiere una atención adecuada. Gracias a los constantes avances de la genética, hoy en día pueden hacerse aclaraciones relevantes desde el punto de vista diagnóstico y terapéutico. Cada vez más personas pueden ser diagnosticadas, confirmadas o descartadas mediante pruebas genéticas con fines médicos (pruebas genéticas) del material genético (ADN), que no deben confundirse con las pruebas genéticas (de estilo de vida) con fines no médicos de Internet o la farmacia (Fig. 1). Además, las aclaraciones médico-genéticas de las enfermedades hereditarias no representan una instantánea del estado de salud, sino que se aplican a lo largo de toda la vida y tienen consecuencias tanto para el paciente como para su familia (parientes consanguíneos). Por ello, la ley exige un asesoramiento genético adecuado por parte de médicos genetistas (GUMG art. 14). El esclarecimiento genético de las enfermedades de manifestación tardía y el estado de portador de las enfermedades recesivas se reserva a los adultos.

Creciente importancia del diagnóstico genético
Tanto en el diagnóstico como en la terapia de las enfermedades debe tenerse en cuenta la influencia de las causas genéticas. La familiaridad puede ser indicativa, pero las enfermedades hereditarias también se dan esporádicamente. En el caso de enfermedades comunes multifactoriales, como la hipertensión, los efectos genéticos suelen ser múltiples y sólo son fuertes en conjunto, mientras que las enfermedades monogénicas están causadas por la mutación de un único gen. A menudo, el cuadro clínico no es claramente pronunciado en tales enfermedades, por lo que la genética contribuye decisivamente al diagnóstico correcto con nuevos métodos y hallazgos. Esta es la base para el pronóstico y la gestión específica de la enfermedad y, si es posible, para el éxito de la terapia y la prevención [1]. Sin un diagnóstico, no se puede hacer ninguna afirmación concluyente sobre la terapia o un cambio de terapia. Por desgracia, este aspecto no suele tenerse en cuenta en nuestro sistema sanitario. Incluso cuando el diagnóstico clínico es aparentemente seguro, la identificación del defecto genético causante de la enfermedad (mutación) puede ser esencial, ya que el tratamiento y las terapias pueden ser específicos del gen o incluso de la mutación.
A diferencia de las pruebas rutinarias rápidas de los parámetros sanguíneos, los exámenes genéticos médicos sólo pueden automatizarse hasta cierto punto y realizarse en un laboratorio de prácticas. Se utilizan principalmente en situaciones en las que los exámenes clínicos no permiten un diagnóstico concluyente. Esto es especialmente importante en enfermedades con un fenotipo clínico superpuesto o inespecífico y en la fase inicial de una enfermedad. Incluso de forma presintomática, puede aclararse si existe o no una predisposición genética a la enfermedad familiar.
Secuenciación de ADN de alto rendimiento
El método más importante para la investigación selectiva de genes es la secuenciación del ADN, que puede determinar la secuencia de las bases nucleotídicas del material genético (A, T, G, C) y detectar así con precisión las mutaciones genéticas. Estos análisis de genes se llevan a cabo con una eficacia sin precedentes gracias a la secuenciación de alto rendimiento (“Next Generation Sequencing”, NGS ). La NGS es más eficaz que el análisis clásico de un solo gen mediante la secuenciación de Sanger y tiene especial éxito en la detección de las causas de las enfermedades, así como en el análisis del ADN libre de células que circula en la sangre y en el examen unicelular de las muestras biológicas más pequeñas.
Con la NGS se examina una combinación seleccionada (el llamado panel) de genes (“secuenciación dirigida”, TS), el genoma completo (“secuenciación del genoma completo”, WGS ; ~3.000 millones de bases de nucleótidos) o su región codificante (“secuenciación del exoma completo”, WES ; ~25.000 genes). Sólo por estas diferencias, no toda la NGS es igual (Fig. 2). Además, existe la diferencia de rendimiento y calidad entre los métodos de NGS, que es relevante para el diagnóstico genético [2].

La TS puede analizar determinadas regiones genéticas de forma especialmente intensiva capturando la secuencia de más de 1.000 copias de ADN (“lecturas de secuenciación”) y detectando así pequeñas cantidades (<1:100) de alelos no de referencia, que están presentes en forma del llamado mosaico. Esto es necesario, por ejemplo, en las pruebas genéticas de los cánceres somáticos. La TS también es barata y, por lo tanto, se utiliza a menudo como primer paso en el cribado de mutaciones. Sin embargo, si la mutación causante de la enfermedad no se encuentra con la ET, la enfermedad permanece sin diagnosticar y el examen debe repetirse con WES o, aún mejor, con WGS. Precaución: Un hallazgo negativo (es decir, ninguna mutación patógena conocida/sin ambigüedad) en los genes examinados no descarta una causa genética de la enfermedad actual.
Retos de los análisis genómicos
Con la NGS, existen limitaciones relevantes para el diagnóstico genético. En primer lugar, la longitud de lectura de la secuencia de la tecnología NGS de Illumina, líder del mercado, es demasiado corta (~150 bases de nucleótidos) para asignar regiones repetitivas/homólogas más largas al genoma de referencia con una posición única [2]. Las últimas tecnologías de secuenciación (por ejemplo, de Pacific Biosciences u Oxford Nanopore Technologies), que pueden leer fragmentos de ADN de varios miles de bases de nucleótidos, prometen ayudar. En segundo lugar, la NGS de regiones de ADN ricas en GC es más difícil porque el par de bases de nucleótidos G y C tiene un enlace más fuerte que el par A y T. Especialmente en TS y WES, las regiones de genes ricas en GC, como suele ocurrir al principio de un gen, no se detectan suficientemente (es decir, con lecturas de secuenciación cubiertos), por lo que a menudo no se cumplen los requisitos de calidad de los diagnósticos genéticos. Con la WGS, este problema se da mucho menos, de modo que la WGS no sólo tiene la ventaja de cubrir la región no codificante (98,5%) del genoma, sino que también cubre mejor que la WES la región codificante de especial importancia clínica (1,5%), especialmente las regiones ricas en GC. La WGS permite así el mejor diagnóstico genético posible de enfermedades hereditarias cuyas causas se desconocen o que están causadas por mutaciones en genes grandes y/o complejos, como los genes causantes de cardiomiopatías DMD (~2,3 millones de bases nucleotídicas) y TTN (364 exones), que son nuestros genes más grandes y más ricos en exones, respectivamente.
El grupo de trabajo del American College of Medical Genetics and Genomics (ACMG) publicó una lista de 59 genes cuyas mutaciones aumentan considerablemente el riesgo de enfermedades aórticas, arritmias y miocardiopatías, entre otras, de modo que es posible e indicado adoptar medidas preventivas [3]. El grupo de trabajo del ACMG recomienda que todos estos genes de riesgo se examinen como parte estándar de los exámenes genéticos exhaustivos, como el WES o el WGS, y que se informe a los pacientes de las desviaciones patogénicas de la secuencia (mutaciones) si así lo desean, para que se pueda iniciar cualquier medida preventiva en una fase temprana (Tab. 1).

La gran cantidad de datos generados en el curso de una WGS puede reducirse al nivel de TS o WES mediante paneles virtuales (in silico) de genes y centrarse en la pregunta clínica [4, 5]. Además, el control de calidad y la interpretación de la cantidad de datos generados por la NGS es un reto complejo, tanto económico como intelectual, que aún no se refleja adecuadamente en la posición actual de la lista de análisis para la NGS. (Fig. 2). Especialmente la recopilación e interpretación de los datos de NGS que son importantes para el diagnóstico requiere muchos conocimientos de genética humana y puede llevar mucho tiempo. Esto se aplica, por ejemplo, cuando las desviaciones de secuencia se encuentran en genes cuyo significado o función aún no se conoce (del todo), como es el caso de aproximadamente la mitad de los genes humanos actuales. Aunque los programas informáticos de interpretación actuales tienen en cuenta muchos parámetros diferentes y proporcionan bases importantes para la evaluación, la interpretación de estas desviaciones de la secuencia (denominadas “variantes de significado desconocido”, VUS) requiere al menos un análisis de segregación en la familia (si es posible) y/o una caracterización funcional correspondiente, lo que a veces resulta complejo (Fig. 2).
Para detectar la desviación de secuencia causante de la enfermedad, son necesarias amplias bases de datos con información vinculada sobre el genotipo y el fenotipo, así como la creación y curación de asociaciones gen-enfermedad y su clasificación. Por ejemplo, el Grupo de Trabajo ClinGen ha categorizado 53 genes que, cuando mutan, pueden causar aneurismas y disecciones hereditarias de aorta torácica, sindrómicas o no sindrómicas, desde “definido/fuerte” hasta “sin pruebas” [6]. Estas categorías o paneles de genes pueden simplificar la búsqueda específica de la causa de la enfermedad, pero requieren un diagnóstico clínico de sospecha preciso y una actualización periódica [5].
Herencia digénica
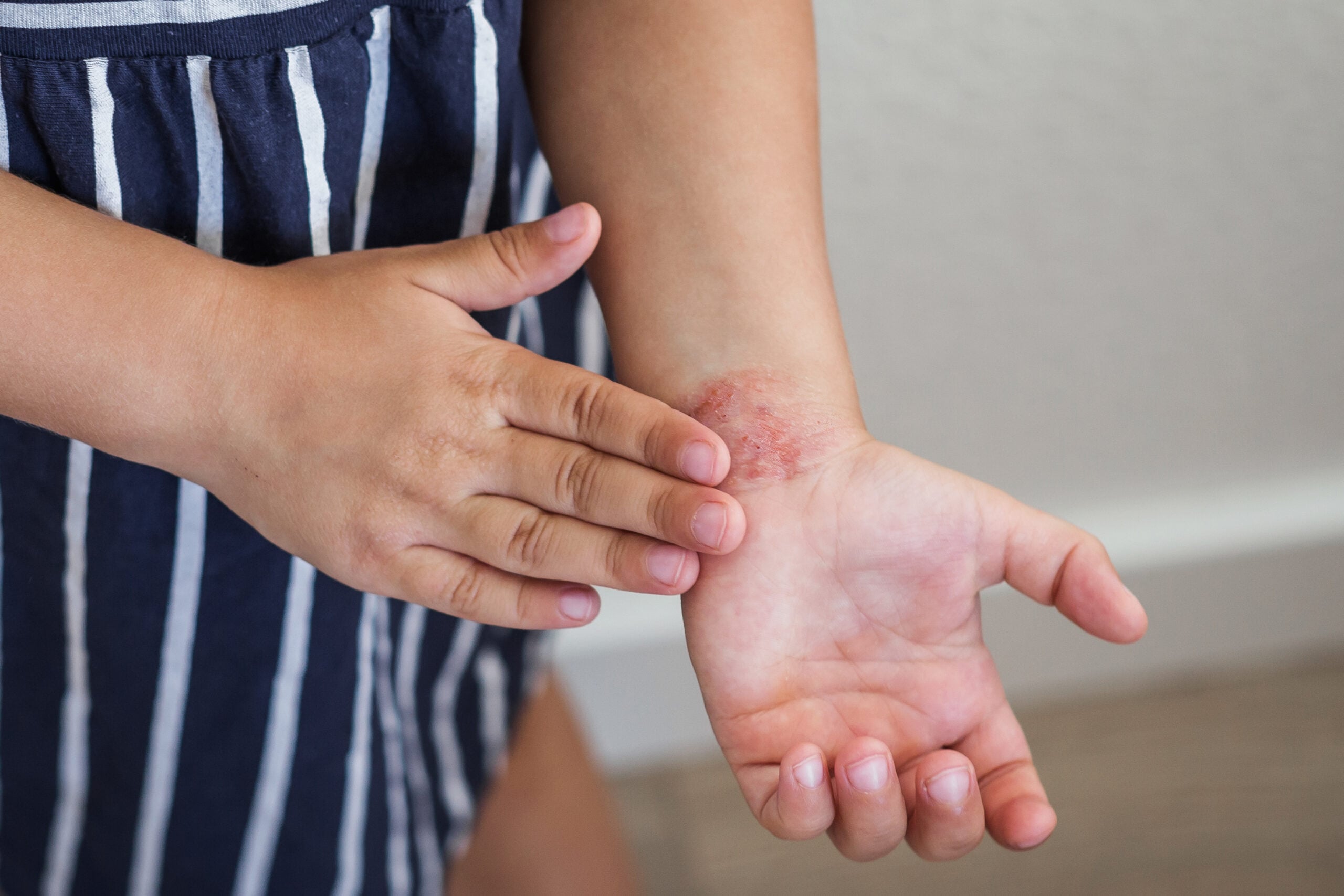
Es poco conocido en la práctica clínica que el cuadro clínico puede basarse en varias enfermedades monogénicas o mutaciones en varios genes que se segregan en la familia y que, por lo tanto, pueden ser importantes en el esclarecimiento genético de los miembros de la familia. A modo de ejemplo, dos familias presentan una mutación en el gen FBN1(asociado al SMF) y/o en el gen FBN2(asociado a la aracnodactilia contractural congénita, CCA) [7]. En aquellos miembros de la familia en los que ambas enfermedades aparecen juntas, no sólo se produce una mezcla sino también una intensificación de los síntomas clínicos, con predominio de los signos clínicos del SMF o de la ECC. Esto es cierto, por ejemplo, en un paciente de 20 años diagnosticado clínicamente de SM clásico (dilatación aórtica con puntuación Z >2, miopía severa, cifoescoliosis) y en comparación con sus padres con signos leves de ACC (padre con FBN2-mutación) o MFS (madre con FBN1-mutación) muestra un fenotipo más pronunciado de SMF (Fig. 3). Igualmente relevante desde el punto de vista clínico es una atenuación o modificación de los síntomas, ya que esto puede hacer que una enfermedad se pase por alto o se diagnostique erróneamente, pero la mutación causante puede seguir siendo hereditaria. La herencia digénica pone de relieve la importancia y la complejidad del esclarecimiento genético.

Del diagnóstico a la terapia
Basándose en un diagnóstico precoz correcto, los ajustes adecuados del estilo de vida, los exámenes de control y/o los tratamientos profilácticos pueden contribuir a la prevención eficaz de enfermedades cardiovasculares como las aortopatías (por ejemplo, el SMF o el síndrome vascular de Ehlers-Danlos, vEDS). La primera opción de tratamiento farmacológico suelen ser los fármacos hipotensores como los betabloqueantes y/o los antagonistas de los receptores de angiotensina II tipo 1 (sartanes). Sin embargo, las propiedades y los efectos tanto de ciertos betabloqueantes (por ejemplo, el atenolol) como de los sartanes (por ejemplo, el losartán) no están exentos de controversia, a pesar de los prometedores estudios (pre)clínicos [8,9]. Esto conduce al uso de diferentes preparados de estas clases de fármacos en función de las preferencias del médico tratante.
Mientras que el losartán se considera el tratamiento farmacológico de elección para prevenir y estabilizar la dilatación aórtica en el SMF, la medicación en el vEDS ha sido incierta, a pesar de un sugerente estudio de The Lancet [10]. La certeza la aporta un trabajo reciente que demostró un claro efecto positivo del betabloqueante celiprolol (Selectol), pero no del losartán, sobre la estabilidad mecánica de la aorta en un modelo experimental de vEDS en ratones [11]. Esto demuestra que el éxito terapéutico de los fármacos antihipertensivos puede variar mucho en función de la aortopatía.
Farmacogenética: el futuro de la farmacoterapia
Los medicamentos se prescriben a menudo según el principio de “uno para todos”, pero no siempre son (igual de) eficaces para todos los pacientes. No sólo influye la mutación causante de la enfermedad, sino también la farmacogenética, es decir, la predisposición genética que influye en el efecto de un fármaco a través de la farmacocinética y la dinámica subyacentes. El campo de la farmacogenética es aún relativamente joven y, a pesar de su importancia no reconocida, sólo se está implantando gradualmente en la práctica clínica diaria. Ya existen perfiles farmacogenéticos de pacientes en Holanda y dentro del proyecto de la UE Farmacogenómica Ubicua (U-PGx). Estos perfiles contienen información sobre las predisposiciones genéticas conocidas más importantes que influyen en el efecto de los fármacos. Junto con los resultados, el paciente recibe una tarjeta en formato de tarjeta de crédito con código QR (Fig. 4), que contiene sus predisposiciones genéticas y la dosis de medicación basada en ellas según las directrices internacionales. En el proyecto U-PGx, se trata actualmente de 45 desviaciones de secuencia en 12 genes y su influencia en el efecto de 76 fármacos. Por ejemplo, para los individuos que son metabolizadores lentos de CYP2D6 (~5-10% de los europeos), se recomienda una reducción de la dosis del betabloqueante metoprolol al 25% de la dosis normal, mientras que para los metabolizadores ultrarrápidos (~3% de los europeos), se recomienda un aumento de la dosis al 250% [12]. Los nuevos hallazgos se integran continuamente y los datos más recientes pueden recuperarse utilizando el código QR del mapa. La tarjeta puede presentarse al visitar a un médico o una farmacia y así la elección o la dosis de un medicamento puede ajustarse individualmente (medicina personalizada o de precisión). Esto no sólo ahorra mucho tiempo de ajuste de la dosis correcta y posibles efectos secundarios (fatales) o ineficacia de los medicamentos en el paciente, sino también costes asociados en el sistema sanitario [13].

Outlook
En el futuro, el diagnóstico genético no sólo será decisivo para el diagnóstico de la mutación causante de la enfermedad y, por tanto, de la terapia (farmacológica) adecuada, sino también para el análisis farmacogenético de los genes que determinan la elección y la dosis de los fármacos utilizados. Los cambios (epi)genéticos que no afectan directamente a la secuencia de ADN de los genes, sino a su regulación, también se estudian y comprenden cada vez más. Cada vez se pueden identificar más efectos genéticos en las enfermedades cardiovasculares multifactoriales comunes.
Nuevos métodos como CRISPR/Cas abren posibilidades, pero también peligros, en el tratamiento de enfermedades genéticas. A pesar de los prometedores resultados de los estudios realizados hasta la fecha, esta tecnología está aún muy lejos de abrirse camino en la práctica médica cotidiana. Por un lado, porque el método aún no está totalmente desarrollado, por otro, porque es necesario regular las cuestiones éticas y jurídicas para limitar cualquier posibilidad de abuso. Sin embargo, es obvio que la genética determinará el futuro de la medicina y que ésta se orientará hacia la genética en la prevención, el diagnóstico y la terapia.
Mensajes para llevarse a casa
- Las pruebas genéticas pueden realizar diagnósticos precisos que de otro modo no serían posibles. Sin embargo, no todas las pruebas genéticas son iguales: El método utilizado determina de forma decisiva la importancia de la prueba.
- La aparición frecuente y/o precoz o sindrómica de una enfermedad en la familia puede indicar una causa genética. Sin embargo, con los conocimientos actuales, la causa genética no puede identificarse en todos los casos.
- El esclarecimiento genético es importante no sólo para el diagnóstico, el pronóstico, la prevención y el asesoramiento familiar, sino también para el tratamiento causal o farmacológico.
- Para el síndrome de Ehlers-Danlos vascular, el celiprolol (Selectol) en lugar del losartán es el tratamiento farmacológico de elección.
Literatura:
- Attenhofer Jost CH, Greutmann M, et al.: Tratamiento médico de los aneurismas aórticos en el síndrome de Marfan y otras afecciones hereditarias. Curr Cardiol Rev 2014; 10(2): 161-171.
- Caspar SM, Dubacher N, et al.: Secuenciación clínica: de los datos brutos al diagnóstico con valor de por vida. Clin Genet 2018; 93(3): 508-519.
- Kalia SS, et al: Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017; 19(2): 249-255.
- Plüss M, Kopps A, et al.: Necesidad de velocidad en el análisis preciso de datos del genoma completo: GENALICE MAP desafía a BWA/GATK más que PEMapper/PECaller e Isaac. Proc Natl Acad Sci U S A 2017; 114(40): E8320-E8322.
- Caspar SM, et al: Más genes para los aneurismas y disecciones de la aorta torácica. J Am Coll Cardiol 2019; 73(4): 528-529.
- Renard M, et al: Validez clínica de los genes de los aneurismas y disecciones hereditarios de la aorta torácica. J Am Coll Cardiol 2018; 72(6): 605-615.
- Najafi A & Caspar SM, et al.: Filtrado de variantes, herencia digénica y otros retos en la era genómica actual: una lección de las fibrilinopatías (presentado).
- Carlberg B, et al: Atenolol en la hipertensión: ¿es una elección acertada? Lancet 2004; 364(9446): 1684-1689.
- Gao L & Chen L, et al: El efecto del losartán sobre la dilatación aórtica progresiva en pacientes con síndrome de Marfan: un metaanálisis de ensayos clínicos prospectivos aleatorizados. Int J Cardiol 2018; 217: 190-194.
- Ong KT, et al: Efecto del celiprolol en la prevención de episodios cardiovasculares en el síndrome de Ehlers-Danlos vascular: un ensayo prospectivo aleatorizado, abierto y ciego. Lancet 2010; 376(9751): 1476-1484.
- Dubacher N & Münger J, et al: El celiprolol pero no el losartán mejora la integridad biomecánica de la aorta en un modelo de ratón del síndrome de Ehlers-Danlos vascular. Cardiovasc Res 2019 (publicación electrónica antes de impresión).
- Swen JJ, et al: Farmacogenética: del banco al byte – una actualización de las directrices. Clin Pharmacol Ther 2011; 89(5): 662-673.
- Verbelen M, et al: Rentabilidad del tratamiento farmacogenético guiado: ¿hemos llegado ya? Pharmacogenomics J 2017; 17(5): 395-402.
CARDIOVASC 2019; 18(3): 12-16