La amiloidosis de transtiretina con miocardiopatía es una enfermedad significativamente infradiagnosticada que a menudo se presenta con síntomas clínicos de insuficiencia cardiaca. Debido a la pirámide de edad, lo más probable es que el número de pacientes aumente. Pero el amplio espectro de síntomas dificulta el diagnóstico. En este contexto, un diagnóstico precoz sería importante para poder aplicar rápidamente la opción de tratamiento existente.
La amiloidosis es un grupo de enfermedades caracterizadas por el depósito de proteínas patológicas en el intersticio. Si el amiloide se forma por la deposición de monómeros de transtiretina (TTR) mal plegados, especialmente en el corazón y los nervios periféricos o autónomos, hablamos de amiloidosis por transtiretina con cardiomiopatía (ATTR-CM). Tiene una elevada morbilidad y mortalidad. El cuadro clínico es tan polifacético que a menudo la ATTR-CM sólo se descubre en fases avanzadas. Por lo tanto, es deseable un estrecho intercambio entre médicos de cabecera, cardiólogos y especialistas en medicina nuclear.
Características típicas
Por ejemplo, en pacientes varones mayores de 60 años con insuficiencia cardiaca con fracción de eyección preservada (IC-FEM), debe considerarse la ATTR-CM si no responden a la terapia estándar con betabloqueantes o inhibidores de la ECA. Un síndrome del túnel carpiano bilateral, el aumento de los valores de NT-proBNP o un engrosamiento de la pared cardiaca superior a 12 mm también son indicativos de esta forma de amiloidosis (tab. 1). Una gammagrafía esquelética puede aportar claridad. El rasgo más característico es un “corazón negro”.

¿De tipo salvaje o hereditario?
La desestabilización de la protreína TTR y la formación de proteínas TTR mal plegadas dan lugar a fibrillas amiloides, que se depositan en distintas partes del cuerpo y pueden provocar graves cambios estructurales en ellas. En la amiloidosis ATTR, los nervios periféricos o el corazón son los principales afectados. En consecuencia, se distingue entre amiloidosis ATTR con polineuropatía o ATTR-CM. La causa puede ser una mutación en el gen de la transtiretina de forma hereditaria o de tipo salvaje relacionada con la edad. La ATTR-CM suele presentarse como tipo salvaje, pero también puede manifestarse de forma mutacional. Se desarrolla de forma insidiosa, pero debe diagnosticarse precozmente. Esto se debe a que la mediana de supervivencia sin tratamiento desde el diagnóstico es de 3,6 años.
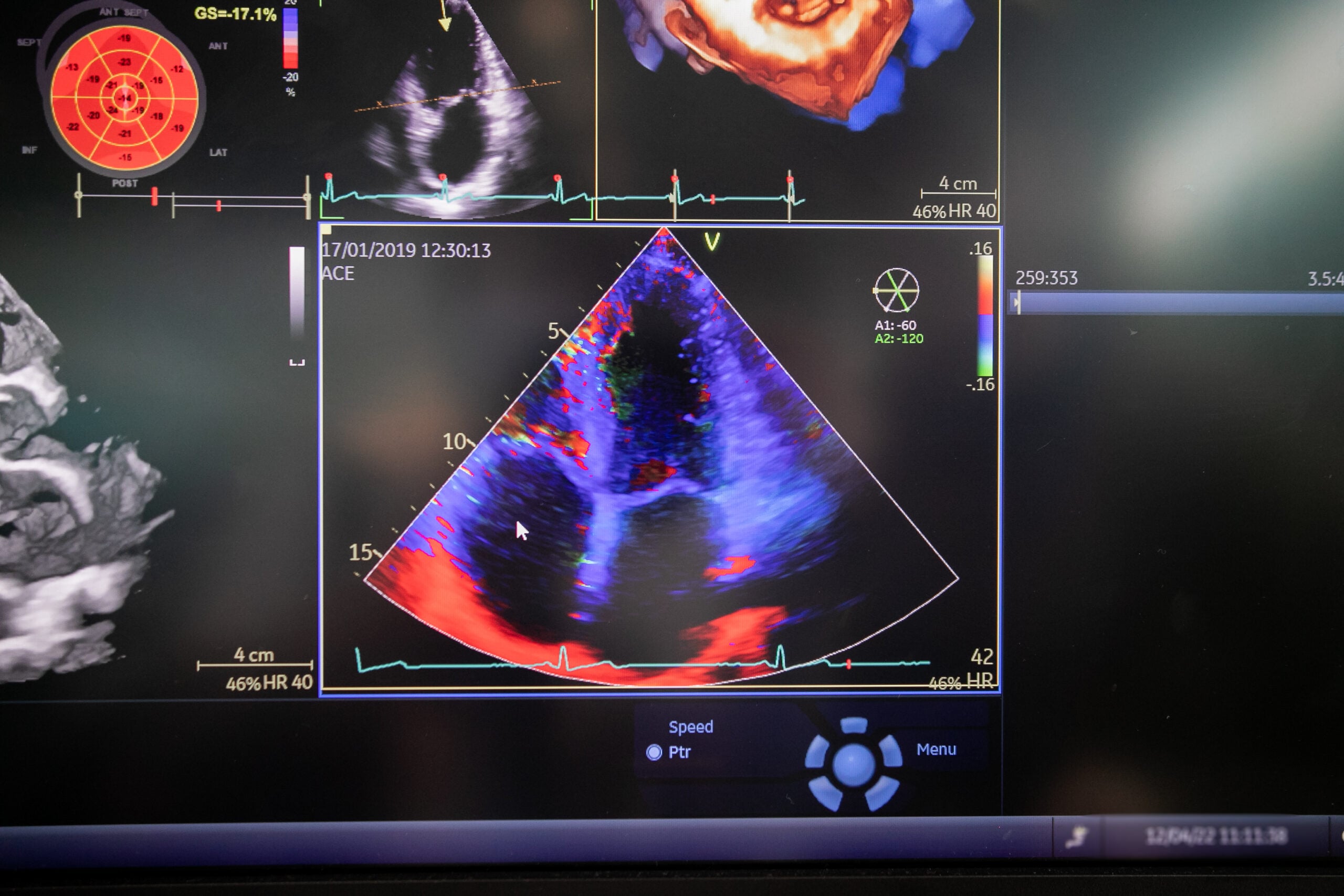
Los diagnósticos se realizan a partir de diferentes medidas. La ecocardiografía puede detectar el engrosamiento de las paredes del corazón. Además, la función de la bomba está limitada y en fases posteriores las aurículas están dilatadas. Una característica especial es la deformación longitudinal casi preservada o sólo ligeramente reducida en los segmentos apicales del ventrículo izquierdo. Para un cribado no invasivo, también se recomienda una gammagrafía esquelética.
Abordar las causas
La primera y hasta ahora única terapia causal disponible es el tafamidis, un inhibidor oral de la disociación del tetrámero TTR. Se une al sitio de unión de la tiroxina del tetrámero de la TTR, lo estabiliza y evita así la formación de fibrillas. En el estudio pivotal, se registró un 30% menos de mortalidad por todas las causas y un 32% menos de hospitalizaciones relacionadas con enfermedades cardiovasculares en los brazos de dosis de 20 mg y 4× 20 mg de meglumina de tafamidis en comparación con el placebo.
Los resultados del estudio de extensión abierto mostraron ahora un beneficio significativo en la supervivencia de los pacientes que recibieron 80 mg de meglumina de tafamidis en comparación con los que recibieron 20 mg, con una reducción relativa del riesgo de muerte del 30%. Cuando se corrigieron las covariables relevantes, la reducción relativa global del riesgo de mortalidad fue del 43%. El fármaco se toleró bien en ambas dosis y el perfil de seguridad fue comparable al del placebo.
Para saber más:
- Maurer MS, et al: Circulation 2017; 135:1357-1377.
- González-López, et al: Eur Heart J. 2015; 36(38): 2585-2594.
- Ruberg FL, et al: J Am Coll Cardiol. 2019; 73(22): 2872-2892.
- Maurer MS, et al: Circ Heart Fail. 2019; 12: e006075
- Donnelly J, Hanna M. Cleve Clin J Med 2017; 84(12 Suppl 3): 12-26.
- Aus dem Siepen, et al: Clin Res Cardiol. 2019; 108(12): 1324-1330.
- Gillmore JD, et al: Circulation 2016; 133: 2404-2412.
- Damy T, et al: Eur J Heart Fail 2020 Oct 18. doi.org/10.1002/ejhf.202
- Yilmaz A, et al: Clinical Research in Cardiology, publ. online enero 2021, https://doi.org/10.1007/s00392-020-01799-3
CARDIOVASC 2021; 20(2): 18