Las vasculitis sistémicas son síndromes inflamatorios de los vasos sanguíneos que pueden causar un espectro muy amplio de síntomas según el calibre del vaso afectado y la localización. Se describen las vasculitis primarias más comunes en adultos con las opciones actuales de tratamiento farmacológico.
Las vasculitis sistémicas son síndromes inflamatorios de los vasos sanguíneos que pueden causar un espectro muy amplio de síntomas según el calibre del vaso afectado y la localización. Dependiendo de la localización, un ataque vasculítico de pequeños vasos, es decir, de capilares, arteriolas y vénulas, puede manifestarse, por ejemplo, como púrpura palpable de la piel, glomerulonefritis necrosante de progresión rápida con insuficiencia renal, hemorragias pulmonares, hemorragias nasales, escleritis o como opacidad cerebral. Si se ven afectadas las arterias medianas y grandes, existe riesgo de infartos tisulares, aneurismas, hemorragias y trombosis. Aunque en los últimos 40 años se han logrado avances significativos en el tratamiento de la vasculitis sistémica, la mortalidad sigue siendo significativamente mayor en comparación con la población general [1]. A continuación se describen las vasculitis primarias más comunes en adultos, junto con las opciones actuales de tratamiento farmacológico.
Clasificación de las vasculitis sistémicas
El grupo de las vasculitis sistémicas “primarias” comprende síndromes de enfermedad “idiopáticos” independientes, mientras que las vasculitis “secundarias” se producen en relación con enfermedades preexistentes. Ejemplos de vasculitis secundarias son la vasculitis crioglobulinémica asociada a la hepatitis C y la vasculitis en la artritis reumatoide seropositiva de larga evolución.
La clasificación de las vasculitis sistémicas primarias según la clasificación de Chapel-Hill se basa en el calibre del vaso afectado [2]. (Visión general1). Las vasculitis de grandes vasos que afectan a la aorta y/o sus ramas incluyen la arteritis de Takayasu, que se da principalmente en mujeres jóvenes asiáticas, y la arteritis de células gigantes (RZA; sinónimo: arteritis temporal), que no suele manifestarse antes de los 50 años. El síndrome de Kawasaki, que afecta a los niños, y la muy rara panarteritis nodosa afectan sobre todo a las arterias de tamaño medio. Las vasculitis primarias de pequeño vaso se dividen en dos grupos principales: Las vasculitis asociadas a ANCA (anticuerpos citoplasmáticos antineutrófilos) y las vasculitis ANCA negativas. Las AAV incluyen la granulomatosis con poliangeítis (GPA; antes enfermedad de Wegener); la poliangeítis microscópica (MPA); la granulomatosis eosinofílica con poliangeítis (EGPA; antes síndrome de Churg-Strauss); y la glomerulonefritis necrotizante focal (FNGN). Varias vasculitis mediadas por inmunocomplejos se asignan a las vasculitis primarias de pequeños vasos ANCA negativas, por ejemplo la vasculitis IgA (Henoch-Schönlein), que se da sobre todo en niños, y la vasculitis crioglobulinémica esencial.

Las vasculitis secundarias suelen afectar a los vasos pequeños (capilares, arteriolas, vénulas) y, al igual que las vasculitis de vasos pequeños ANCA negativas, suelen estar causadas por complejos inmunitarios con activación del sistema del complemento. El tratamiento de la vasculitis secundaria siempre está relacionado con la enfermedad subyacente y muy a menudo implica el uso a corto plazo de dosis más altas de glucocorticoides y, en algunos casos (por ejemplo, la vasculitis crioglobulinémica inducida por virus), el agotamiento terapéutico de los linfocitos B CD20+ [3]. La enfermedad de Behçet ocupa una cierta posición especial entre los síndromes de vasculitis, en los que pueden verse afectados todos los calibres de vasos.
En todas las vasculitis, el primer objetivo del tratamiento es lograr una remisión clínica lo más rápida posible, lo que se denomina inducción de la remisión. A ésta le sigue la fase de mantenimiento de la remisión, que puede durar varios años según el tipo de vasculitis.
Arteritis de células gigantes (RZA)
La RZA, también conocida como arteritis temporal, es la vasculitis más común en nuestras latitudes y suele aparecer después de los 60 años, y en la gran mayoría de los casos después de los 50. Afecta a la aorta y a las arterias grandes y medianas que se ramifican de ella (Fig. 1). Los síntomas típicos son nuevas cefaleas, a menudo “neurálgicas”, un síndrome polimiálgico, sintomatología B con descenso del rendimiento, agotamiento y sudores nocturnos, síntomas de claudicación de las extremidades, la lengua y/o los músculos masticatorios, así como alteraciones visuales. La RZA es temida sobre todo por el peligro de ceguera aguda, que suele ser irreversible. En términos de laboratorio, es típica -pero no obligatoria- una reacción de fase aguda pronunciada, con una reacción de sedimentación marcadamente acelerada (BSR) y una elevación de la proteína C reactiva (CRP).
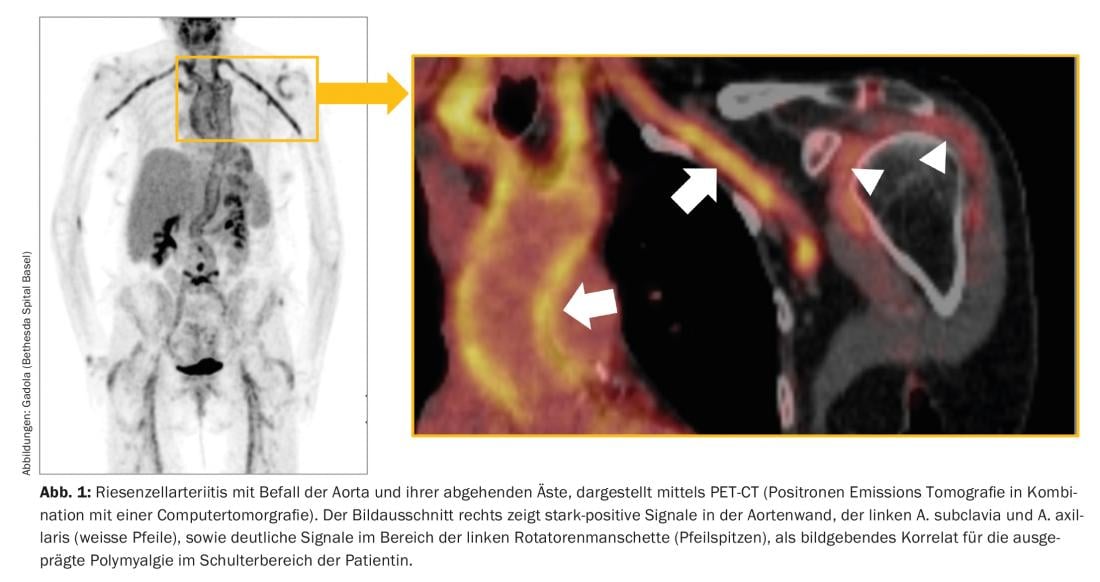
Los glucocorticoides son los fármacos de primera línea para el RZA y suelen producir una mejora drástica de los síntomas subjetivos en 24 horas. Si se sospecha una afectación ocular, por ejemplo en caso de alteraciones transitorias de la agudeza visual, y una posible ceguera inminente, se administran dosis elevadas por vía intravenosa, por ejemplo 1 g de metilprednisolona en tres días consecutivos. En otros casos, una dosis diaria inicial de 40-60 mg es suficiente. La dosis se ajusta según la clínica y los valores de laboratorio. Mientras la dosis de prednisona no pueda reducirse por debajo de 20 mg/día, debe administrarse profilaxis antibiótica con sulfometoxazol-trimetoprima (por ejemplo, Cotrim-CT 800/160 mg; Bactrim forte®) 3×1/semana para prevenir las infecciones oportunistas, especialmente las causadas por Pneumocystis jirovecii.
En el pasado, se utilizaron dosis de prednisona de 1 mg/kg de peso corporal durante todo un año en la RZA, al igual que en otras vasculitis, con frecuente aparición de efectos secundarios graves. Sin embargo, con la ayuda de fármacos antirreumáticos básicos ahorradores de esteroides (fármacos antirreumáticos modificadores de la enfermedad, FAME), la dosis de prednisona puede reducirse mucho antes por debajo de la llamada dosis umbral de esteroides de 7,5 mg de prednisona. El metotrexato (MTX) en particular ha demostrado ser un buen DMARD para la RZA. El inicio de la acción se retrasa con el MTX, como con todos los DMARD, y se produce aproximadamente entre 4 y 6 semanas después de alcanzar la dosis eficaz.
El metotrexato debe administrarse siempre por vía parenteral, es decir, subcutánea (s.c.) una vez a la semana, para el tratamiento de las vasculitis. Si se tolera bien, la dosis puede aumentarse hasta un máximo de 0,3 mg/kgKG por semana. La BSR y la PCR también sirven como valiosos biomarcadores de progresión de la actividad inflamatoria bajo tratamiento con prednisona y DMARD. Para reducir los efectos secundarios tóxicos del MTX, el ácido fólico, por ejemplo 10 mg/semana, debe tomarse siempre de forma concomitante al día siguiente de la inyección de metotrexato. El MTX no debe combinarse nunca con sulfometoxazol-trimetoprima, ya que de lo contrario podría producirse una mielosupresión grave. Por lo tanto, sólo utilizamos MTX después de que la dosis de prednisona se haya reducido a menos de 20 mg/día.
Antes de iniciar una terapia básica ahorradora de esteroides con MTX u otros DMARD, debe realizarse una radiografía de tórax para excluir una infección crónica o fibrosis pulmonar, un hemograma diferencial, valores hepáticos y renales, y pruebas serológicas de hepatitis B, hepatitis C y VIH. Durante los 3 primeros meses bajo metotrexato, se recomiendan controles mensuales de los valores hepáticos y renales y del recuento sanguíneo; después, estos controles pueden realizarse a intervalos más largos, de 8 a 12 semanas, si es necesario. En los portales de Internet de las sociedades reumatológicas se pueden encontrar recomendaciones detalladas sobre el uso de los DMARD en las enfermedades reumatológicas [por ejemplo, 4].
Desde 2017 (zona UE) resp. 2018 (Suiza), se aprueba el anticuerpo contra el receptor de la interleucina 6 (anti-IL6R) tocilizumab (Actemra®) para el tratamiento del RZA. Con el tocilizumab, la dosis de glucocorticoides puede reducirse mucho más rápidamente, incluso sin metotrexato, con un buen éxito clínico [5,6]. La terapia con tocilizumab debe llevarse a cabo durante al menos 1 año, ya que de lo contrario las recidivas son frecuentes [7]. La BSR y la PCR se suprimen con tocilizumab y, por tanto, no son útiles como parámetros de progresión de la actividad de la enfermedad. Datos recientes sugieren que la dosis de glucocorticoides bajo tocilizumab puede interrumpirse muy rápidamente, es decir, en pocas semanas [8]; sin embargo, se necesitan más estudios antes de poder hacer recomendaciones claras. Dado que actualmente los glucocorticoides en la RZA no pueden reducirse por debajo de la dosis umbral durante un periodo de tiempo prolongado (>3 meses), se recomienda el inicio rápido de una terapia antirresortiva, por ejemplo con alendronato, para prevenir la resorción ósea o la pérdida de hueso adquirida por los esteroides. de la osteoporosis.
Arteritis de Takayasu (AT)
Al igual que la RZA, la AT afecta a la aorta y a las grandes arterias que parten de ella. A diferencia de la RZA, esta vasculitis se da en un 80-90% de los casos en mujeres, con un inicio entre los 10 y los 40 años . La AT progresa en episodios y se manifiesta típicamente con síntomas constitucionales, artralgias y, de forma característica, un marcado dolor a la presión carotídea (en el 10-30%). En el curso de la enfermedad pueden producirse oclusión vascular, hipertensión renovascular grave, retinopatía (de Takayasu) y aneurismas aórticos con o sin insuficiencia de la válvula aórtica.
Cuando se diagnostica una AT, en primer lugar se utilizan glucocorticoides. Los FAME modificadores de la enfermedad ahorradores de esteroides más utilizados en la AT son el metotrexato s.c. (como en la RZA) o la azatioprina p.o. (hasta 2 mg/kgKG). Las alternativas son el micofenolato (1,5 g-3 g/día p.o.) y la leflunomida (20 mg/día p.o.). En casos de intolerancia al metotrexato o a los DMARD orales, casos resistentes a la terapia y casos graves, se utilizan bloqueadores del TNFalfa (por ejemplo, etanercept o infliximab) u otros productos biológicos (por ejemplo, tocilizumab, abatacept, ustekinu-mab), pero aún no están aprobados para esta indicación [9].
Vasculitis asociadas a ANCA (AAV)
GPA (antes enfermedad de Wegener)
La GPA es la vasculitis primaria de pequeño vaso más frecuente en nuestras latitudes, con una distribución por sexos casi equilibrada y una edad típica de manifestación entre los 40-60 años. Año de vida. Se distingue entre una “fase inicial” no vasculítica y granulomatosa (localizada) y una “fase de generalización” sistémica y vasculítica, que pueden producirse de forma secuencial (fase inicial ‘ fase de generalización) o simultánea. Aunque hoy en día se dispone de una amplia gama de terapias eficaces para el tratamiento de la vasculitis de pequeños vasos, el tratamiento de las manifestaciones inflamatorias granulomatosas agresivas suele ser un reto importante [10,11].
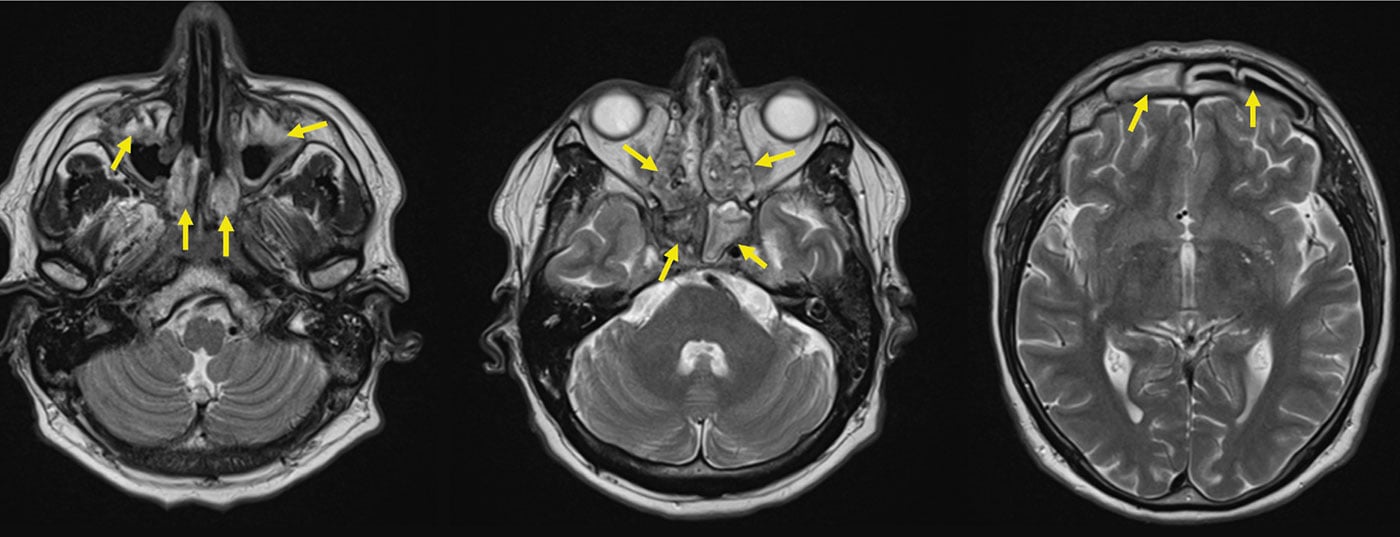
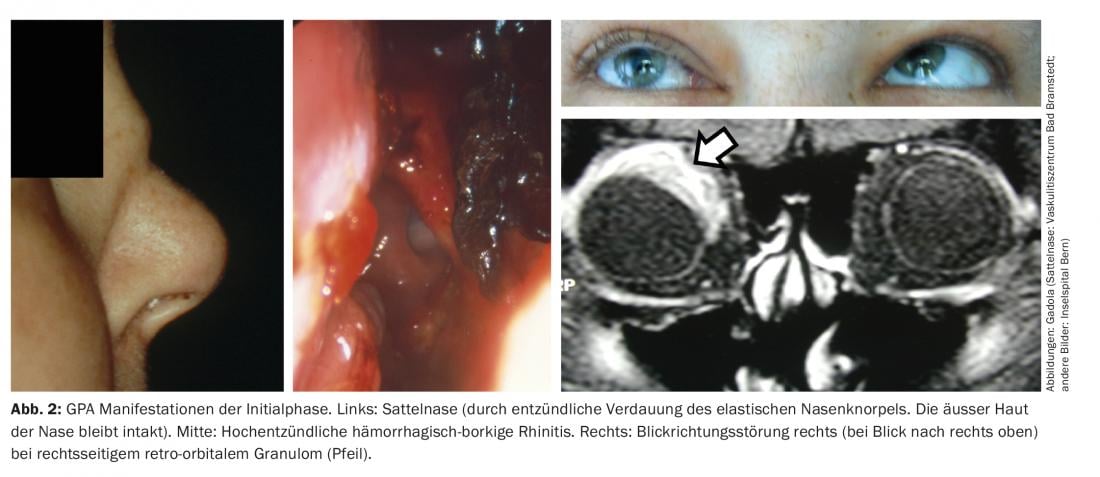
Fase inicial de la GPA (GPA “localizada”). La fase inicial de la GPA suele manifestarse en el tracto auditivo, nasal y faríngeo y en las vías respiratorias, por ejemplo en forma de rinitis borkiana hemorrágica crónica, sinusitis resistente a la terapia o mastoiditis. En un curso agresivo, puede producirse una nariz en silla de montar, debido a la destrucción del cartílago nasal elástico, la formación de fístulas hacia la órbita o hacia la cara, granulomas retroorbitarios con alteraciones de la dirección de la visión (diplopía) y paquimeningitis granulomatosa (Fig. 2). Las dos últimas se deben probablemente a una inflamación granulomatosa originada en los senos per continuitatem [11]. Los ANCA con especificidad para la proteinasa-3 (PR3-ANCA) o, más raramente, la mieloperoxidasa (MPO-ANCA) sólo son detectables en aproximadamente el 50% de los pacientes con GPA durante la fase inicial.
Para el tratamiento de la fase inicial “pura” sin vasculitis sistémica concomitante, se utilizan diferentes combinaciones de fármacos en función de la gravedad de los síntomas. En los casos más leves, se utiliza sulfometoxazol-trimetoprima (T/S) con o sin dosis bajas de prednisona (10 mg/día). El uso de la T/S se remonta a una observación empírica realizada en la década de 1970 por Richard Deremee en la Clínica Mayo [12]. En la década de 1990, se demostró una asociación entre la colonización nasal crónica por Staphylococcus aureus y la actividad de la enfermedad en la GPA [13]. Desde entonces, también se ha utilizado el tratamiento antibiótico tópico intranasal con mupirocina, pero sin un éxito rotundo. Un análisis reciente del microbioma endonasal en la GPA ha mostrado una interesante asociación de la actividad de la enfermedad en la GPA con el Corynebacterium tuberculostearicum [14]. Este patógeno es un patógeno importante en otras enfermedades granulomatosas [15]. El Corynebacterium tuberculostearicum es resistente a la mayoría de los antibióticos [16], lo que puede explicar el éxito moderado de la T/S y la mupirocina.
En el curso más agresivo de la fase inicial, el metotrexato (siempre sin T/S, ya que de lo contrario se combinaría toxicidad para la médula ósea) se utiliza principalmente en combinación con prednisona. En los casos refractarios, los grandes granulomas pulmonares y también los granulomas retroorbitarios, la terapia con anticuerpos anti-CD20 (Rituximab) puede ser eficaz [17]. Los granulomas en la GPA tienen una alta densidad de células B CD20+ y se consideran un posible lugar de origen de la producción de ANCA [18,19]. Una manifestación particular de la GPA durante la fase inicial es la estenosis traqueal fibrosante inflamatoria con disnea y estridor inspiratorio, que puede requerir tratamiento con infiltración local de glucocorticoides y dilatación con balón.
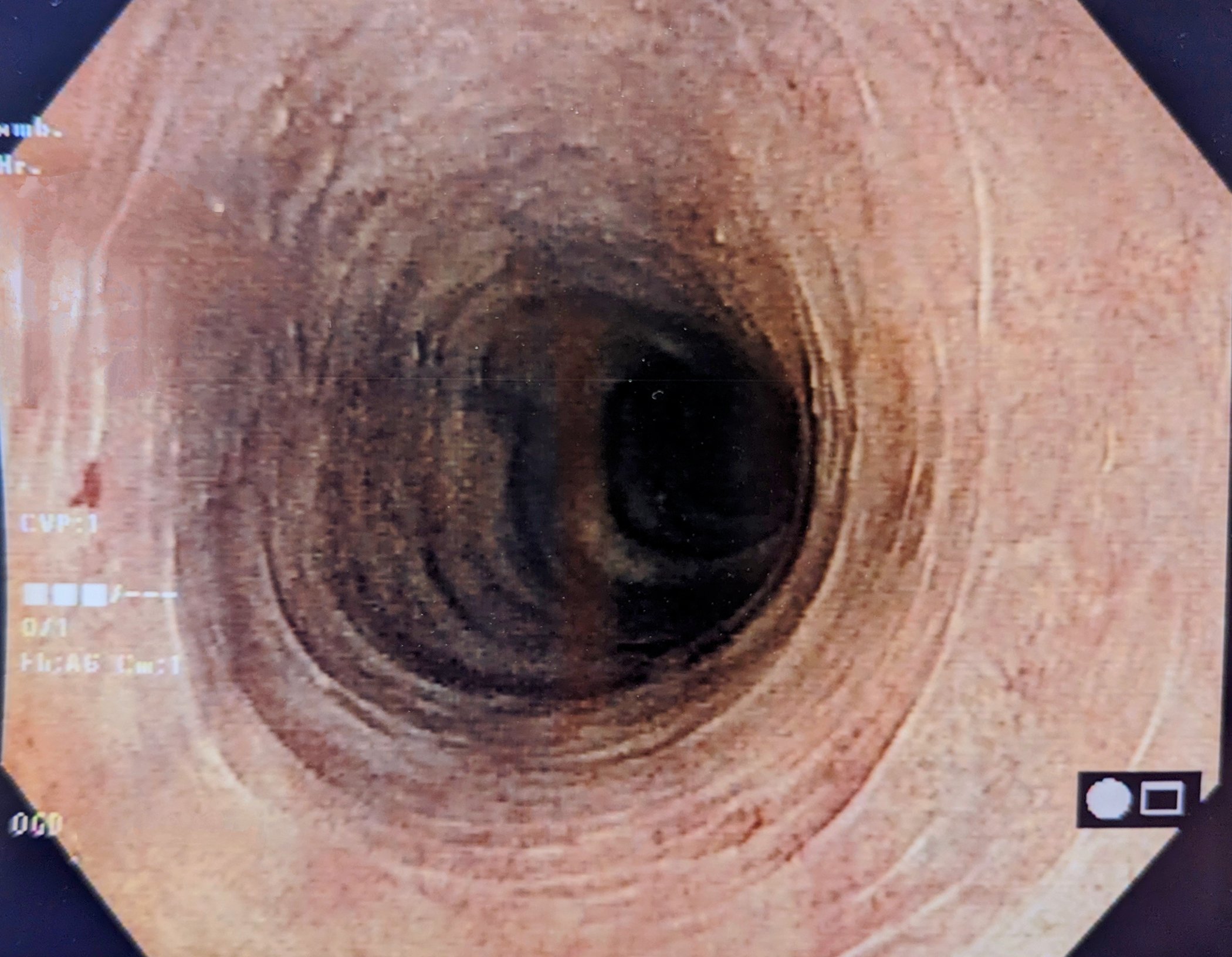
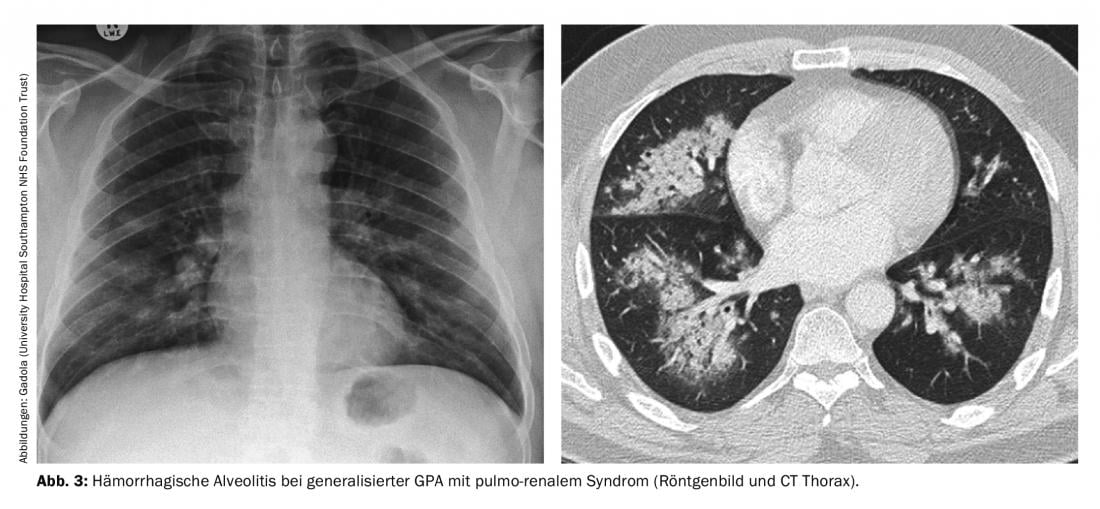
Fase generalizada de GPA (ANCA en >98%). La vasculitis sistémica de pequeños vasos en la GPA, en la que prácticamente siempre puede detectarse PR3-ANCA o MPO-ANCA en el suero, puede afectar a todos los órganos. La infección de los riñones suele dar lugar a una glomerulonefritis “pauciinmune” (RPGN) rápidamente progresiva y necrotizante que, si no se trata, puede desembocar rápidamente en una insuficiencia renal grave que requiera diálisis. El síndrome pulmonar, o hipertensión pulmonar, es especialmente temido. la aparición combinada de RPGN con alveolitis hemorrágica (Fig. 3) , que tiene una elevada mortalidad. Otras manifestaciones típicas de la fase de generalización son una clara sintomatología B, la escleritis -típica es la escleritis nodular dolorosa- que puede desembocar en escleromalacia, la mononeuritis múltiple a menudo muy dolorosa, la encefalitis con opacificación, la púrpura palpable y la poliartritis no erosiva. Los siguientes principios de tratamiento para la GPA generalizada pueden aplicarse a la MPA y la FNGN, y parcialmente a la EGPA.
Inducción de remisión en la GPA generalizada. Dependiendo de la gravedad de la manifestación, se dispone de diferentes modos de acción para inducir la remisión. La regla principal en la GPA generalizada es que la actividad inflamatoria debe controlarse siempre de la forma más rápida y completa posible.
En el caso de una GPA sistémica no renal y que, por lo demás, no ponga en peligro ningún órgano, la terapia de inducción de la remisión puede iniciarse con metotrexato más prednisona o con pulsos de ciclofosfamida intravenosa (i.v.) (por ejemplo, 4 pulsos de 10 mg/kgKG cada uno a intervalos de 3 semanas) más prednisona, siempre que sea posible un estrecho seguimiento de la evolución clínica al menos una vez a la semana. En el caso de las manifestaciones renales y otras que pongan en peligro los órganos, además de la prednisona, se utiliza principalmente la ciclofosfamida por vía oral (empezando con 2 mg/kgKG/día) o en forma de pulso i.v., o el rituximab (2 infusiones i.v. con 1g de rituximab cada una a intervalos de 14 días) en combinación con la prednisona. La elección entre el tratamiento con ciclofosfamida oral, más intensivo en dosis, y la terapia de pulso requiere siempre una cuidadosa consideración de la relación riesgo-beneficio por parte del especialista experimentado en ello. Bajo ciclofosfamida, deben determinarse con regularidad los leucocitos en particular, y especialmente los granulocitos neutrófilos, que suelen aumentar significativamente en las vasculitis GPA activas. Los leucocitos deben alcanzar el nadir en 8-10 días bajo ciclofosfamida, que siempre debe determinarse. Si los leucocitos no descienden, esto indica una actividad persistente de la enfermedad, lo que puede hacer necesario el aumento “adaptado a los leucocitos” de la dosis a 2,5-3 mg/kgKG. Bajo la ciclofosfamida, pueden producirse infecciones oportunistas graves poco después del inicio de la terapia, cuando se produce la leucopenia, especialmente la reactivación del citomegalovirus (CMV). Los efectos secundarios tóxicos típicos de la vejiga relacionados con la dosis son la cistitis hemorrágica y los carcinomas vesicales, que se producen tras una dosis acumulada de al menos 25 g (y una media de 100 g) de ciclofosfamida [20], así como el síndrome mielodisplásico (SMD). La probabilidad de complicaciones vesicales puede reducirse mediante una buena hidratación durante todo el periodo de tratamiento y el uso adicional de Mesna (Uromitexan), que se une y neutraliza el metabolito acroleína de la ciclofosfamida.
Con el rituximab puede producirse un agotamiento duradero de las células B con inmunodeficiencia humoral secundaria e infecciones respiratorias graves frecuentes, especialmente en pacientes que hayan recibido previamente ciclofosfamida [21].
En cursos especialmente graves y/o refractarios de GPA sistémica, a veces se utilizan medidas y fármacos adicionales, como la plasmaféresis, la administración intravenosa de inmunoglobulinas, el micofenolato mofetilo (MMF), el anticuerpo anti-CD52 depletor de células B y T alemtuzumab, la globulina antitimocítica, la 15-deoxispergualina -un inhibidor de la diferenciación celular- y el trasplante de células madre hematopoyéticas. El éxito de estos tratamientos intensificados no está bien establecido y su uso debería restringirse a centros experimentados.
Un nuevo mecanismo de acción muy prometedor para el tratamiento de la GPA generalizada, así como de otras AAV, es la inhibición del sistema del complemento. El avacopan, un antagonista oral del receptor C5a, ha mostrado resultados muy prometedores en ensayos de fase 3, por lo que la probabilidad de aprobación en un futuro próximo es alta. En los pacientes con GPA generalizada que recibieron Avacopan además de la terapia estándar (con ciclofosfamida o rituximab), los glucocorticoides pudieron suspenderse muy rápidamente. Curiosamente, la función renal en los pacientes tratados con Avacopan mostró una mejora continua a lo largo del periodo de tratamiento de 52 semanas [22].
Un fármaco existente para inhibir el sistema del complemento es el anticuerpo anti-C5 eculizumab (Soliris®), que se ha utilizado esporádicamente para la progresión agresiva de la AAV [por ejemplo, 24]. Lamentablemente, un ensayo de fase 2 de eculizumab en vasculitis ANCA se retiró antes de que se inscribieran pacientes (NCT01275287). Otro fármaco, el Iptacopan (LNP023), que inhibe la activación eficaz del sistema del complemento a través del factor B, ha recibido recientemente la aprobación para el tratamiento de la glomerulopatía C3 (C3G), y es muy posible que este fármaco también se utilice en la AAV en el futuro.
Mantenimiento de la remisión en la GPA generalizada. Una vez alcanzada la remisión clínica, la terapia de mantenimiento de la remisión debe salvaguardar la actividad inflamatoria de la AAV incluso con pequeñas dosis de prednisona o incluso sin glucocorticoides concomitantes. Desde 1995, el Grupo Europeo de Estudio de la Vasculitis (EUVAS) ha llevado a cabo un gran número de estudios de intervención clínica en la AAV. Se recomienda MTX, azatioprina o rituximab para mantener la remisión en la GPA y otras AAV. El rituximab es posiblemente el fármaco más eficaz de los tres, aunque la dosis y el intervalo de dosis óptimos del rituximab en la AAV siguen siendo objeto de debate. Algunos expertos recomiendan un intervalo fijo de 6 meses de 1g de rituximab [24], mientras que un estudio comparativo francés de rituximab frente a azatioprina mostró buenos resultados para un intervalo de 6-12 meses con dosis de 500mg [25]. Con rituximab, al igual que con ciclofosfamida, los niveles de PR3 o MPO-ANCA caen por debajo del límite de detección en la mayoría de los casos, correlacionándose con la remisión clínica de la vasculitis. En estos pacientes, según nuestra experiencia, el intervalo de rituximab puede ajustarse al rebote de los títulos ANCA, ampliando el intervalo a más de 12 meses en algunos casos.
Otros AAV (MPA, FNGN, EGPA)
AMP y FNGN
A diferencia de la GPA, en la MPA y la FNGN no se producen granulomas. Estos AAV se asocian ligeramente con más frecuencia a la MPO-ANCA que a la PR3-ANCA, pero esto no tiene importancia para el tratamiento. Ambas AAV se manifiestan típicamente como glomerulonefritis progresiva rápida “pauciinmune” (RPGN). El término “pauciinmune” hace referencia a la detectabilidad débil o ausente de depósitos de inmunoglobulinas en el examen inmunohistoquímico de las biopsias renales. Otras manifestaciones típicas de la MPA son la mononeuritis múltiple dolorosa y la alveolitis, que puede conducir a una fibrosis pulmonar con un aumento significativo de la mortalidad en aproximadamente un tercio de los pacientes [26]. El tratamiento de la MPA y la FNGN es análogo a los principios de tratamiento descritos anteriormente para la GPA generalizada.
EGPA (antes: síndrome de Churg-Strauss/vasculitis)
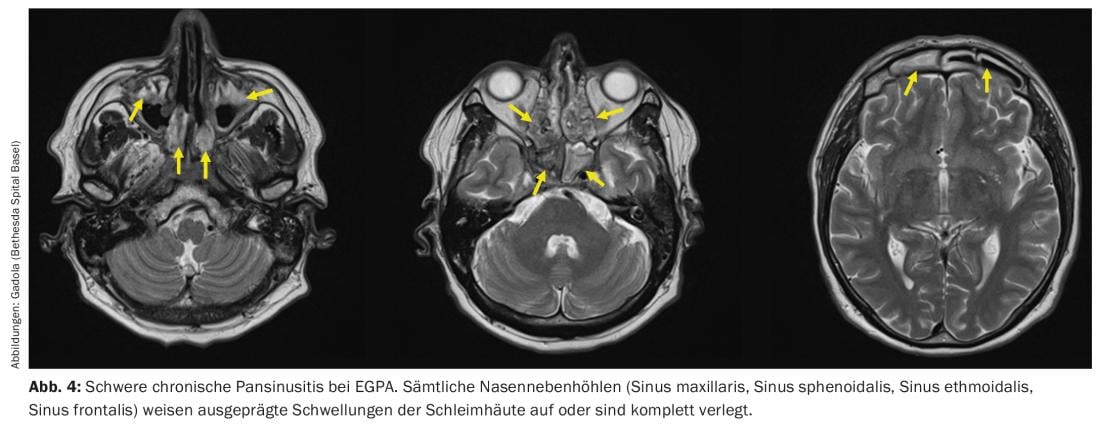
El EGPA puede describirse como el “homólogo atópico” del GPA. Se da en pacientes con síntomas atópicos presentes desde hace años o décadas, especialmente asma y/o pansinusitis crónica (Fig. 4) con pólipos nasales. Durante la fase inicial de la EGPA, se encuentra un granuloma en el tejido afectado, similar al de la GPA. Sin embargo, a diferencia de la GPA, estos granulomas no están densamente entremezclados con granulocitos neutrófilos, sino con granulocitos eosinófilos. En la sangre periférica de los pacientes con EGPA se encuentra análogamente una eosinofilia. El EGPA difiere clínicamente del GPA en aspectos importantes. La aparición de la GNRP es con aprox. un 15-20% más rara que en la GPA [27], pero al igual que en la GPA está estrictamente asociada a la presencia de ANCA. Las manifestaciones neurológicas, especialmente una mononeuritis múltiple a menudo muy dolorosa [28], se producen en más de la mitad de los pacientes. Las manifestaciones más temidas de la EGPA son las cardiacas, por ejemplo la vasculitis de las arterias coronarias, que se dan en cerca del 40% de los pacientes y son la causa más común de muerte en la EGPA [29,30].
La terapia de la EGPA es muy similar a la de la GPA generalizada, y los fármacos más comunes en la EGPA, al igual que en la GPA, son los glucocorticoides, el MTX, la ciclofosfamida y el rituximab. Las dosis altas de glucocorticoides provocan muy rápidamente un descenso drástico de los eosinófilos y una mejoría clínica. Sin embargo, como en el caso de la GPA, el tratamiento con esteroides debe reducirse rápidamente y a la dosis umbral de prednisona en un plazo de 3 meses para reducir el riesgo de infecciones graves de las vías respiratorias. En el caso de la EGPA recidivante o resistente a la terapia, también se dispone de interferón alfa (de 3×/semana a administración diaria s.c.) y del anticuerpo anti-IL5 mepolizumab (Nucala®, 300 mg s.c. cada 4 semanas), que también está aprobado para esta indicación. Con una terapia eficaz, la eosinofilia debería desaparecer y el título de ANCA debería disminuir.
Vasculitis de pequeños vasos no asociada a ANCA
Vasculitis crioglobulinémica esencial
Esta vasculitis de pequeños vasos está causada por la activación in situ de la cascada del complemento en las paredes de los vasos tras la deposición de crioglobulinas de tipo II (menos comúnmente de tipo III). Se manifiesta principalmente en la piel con vasculitis urticarial o púrpura palpable (Fig. 5) . La afectación renal suele presentarse con un complejo inmunológico similar al lupus y una glomerulonefritis membranoproliferativa mediada por complemento. Otros síntomas típicos son mialgias y artralgias, así como polineuropatía. Durante las recaídas, los complementos C4 (“siempre”) y C3 (“a menudo”) disminuyen en el suero.
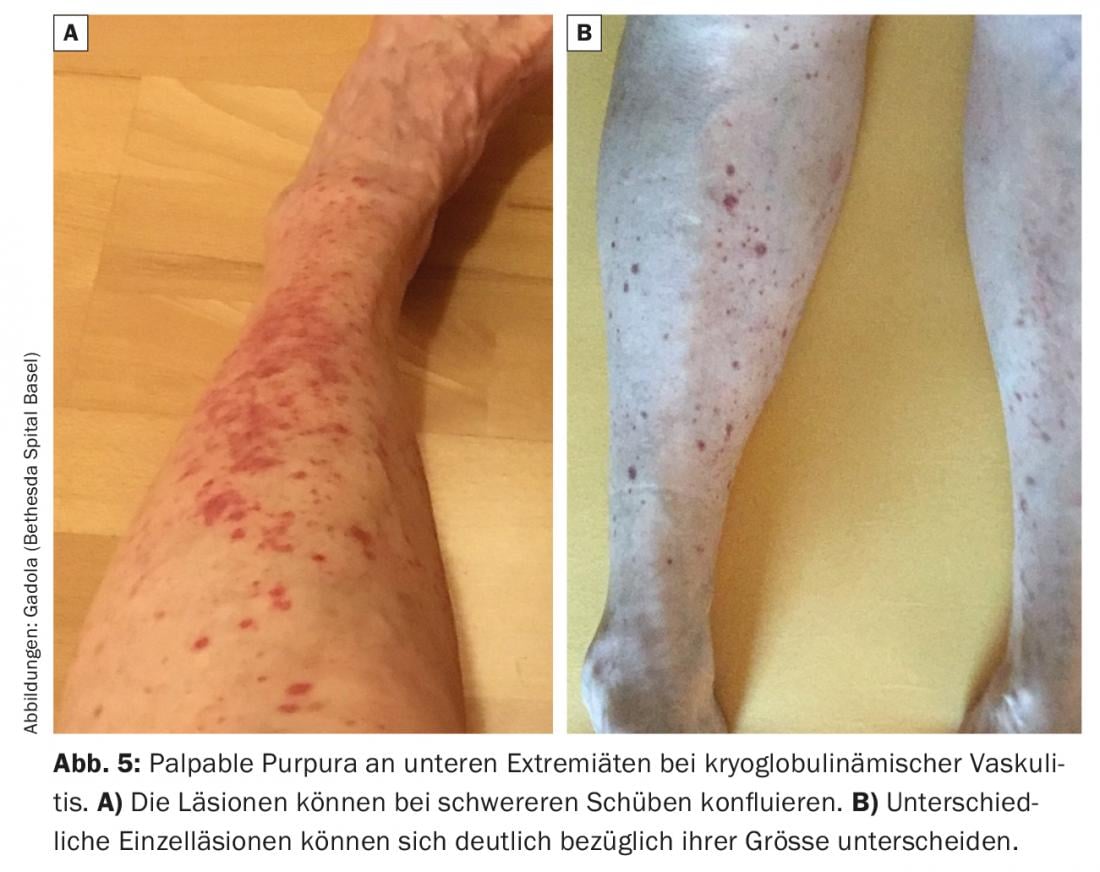
La terapia comienza con glucocorticoides y se complementa con DMARD (MTX o azatioprina) si es necesario. En los casos refractarios, el rituximab ha demostrado ser muy eficaz, y en los casos agudos graves con afectación renal severa, se utiliza la ciclofosfamida [31].
Más comunes que la vasculitis crioglobulinémica esencial, en la que la etiología es por definición desconocida, son la vasculitis crioglobulinémica asociada al VHC (crioglobulinas tipo II o tipo III en suero) y los episodios de vasculitis crioglobulinémica en la neoplasia de células B (crioglobulinas tipo I o tipo III). Aquí, el tratamiento de la enfermedad subyacente ocupa el primer plano, así como el tratamiento depletor de células B con rituximab en los casos graves.
Vasculitis leucocitoclástica
Por lo general, la vasculitis leucocitoclástica mediada por complejos inmunes se limita estrictamente a los capilares y vénulas de la dermis y se manifiesta con púrpura palpable de pruriginosa a dolorosa de las extremidades inferiores. Si hay otros órganos afectados además de la piel, siempre debe buscarse otra vasculitis de pequeños vasos primaria o secundaria. Vasculitis leucocitoclástica confinada a la piel. suele ser autolimitada; sin embargo, también pueden producirse cuadros recurrentes o crónicos graves, que se tratan con glucocorticoides y, si es necesario, con diversos DMARD; en casos especialmente graves, también con ciclofosfamida.
Otras vasculitis “idiopáticas” (y por tanto “primarias”) de pequeños vasos del adulto son la vasculitis urticarial hipocomplementémica (HUV) asociada a anticuerpos anti-C1q, la vasculitis IgA (Henoch-Schönlein) y la vasculitis anti-MBG (GBM, membrana basal glomerular). La terapia de estas vasculitis corresponde en gran medida a los principios descritos anteriormente para otras vasculitis de pequeño vaso.
Enfermedad de Behçet
La enfermedad de Behçet se caracteriza por un amplio espectro clínico y un curso recidivante. La vasculitis en la enfermedad de Behçet puede afectar a vasos arteriales y/o venosos de todos los calibres. La vasculitis de las arterias pulmonares con formación consecutiva de aneurismas y ruptura en el tejido pulmonar es la causa directa de muerte más frecuente. También se producen complicaciones graves por trombosis venosa inflamatoria, afectación cardiaca y cerebral.
La enfermedad de Behçet presenta algunas similitudes clínicas sorprendentes con la enfermedad de Crohn, como la presencia de colitis, formaciones de fístulas enterocolíticas y perianales, aftas mucocutáneas, artritis, eritema nodoso y uveítis. La terapia de las manifestaciones gastrointestinales en la enfermedad de Behçet así lo refleja: además de los glucocorticoides, se utilizan el ácido 5-aminosalicílico (5-ASA), la azatioprina y, si la respuesta es insuficiente, los bloqueantes del TNFalfa. Dependiendo de la gravedad, pueden utilizarse esteroides tópicos, colchicina o el inhibidor de la fosfodiesterasa-4 apremilast (Otezla®), que está aprobado para este fin, para la terapia de las aftas mucocutáneas. En la afectación ocular aguda, se recomiendan principalmente los glucocorticoides sistémicos (por ejemplo, 1 g de metilprednisolona i.v. para la uveítis hipopiónica), siempre en combinación con un FAME, ante todo ciclosporina A o azatioprina, o con interferón-alfa o un bloqueante del TNFalfa (infliximab o adalimumab) [32]. Para la afectación vasculítica de las arterias pulmonares, los glucocorticoides a dosis altas en combinación con ciclofosfamida o bloqueantes del TNFalfa son eficaces. También se utilizan intervenciones no farmacológicas para la enfermedad de Behçet grave. Cuando existe una amenaza de hemorragia por un aneurisma de arteria pulmonar de gran tamaño, se recomienda principalmente el tratamiento de embolización en lugar de la revisión quirúrgica torácica abierta del vaso. En caso de hemorragia gastrointestinal grave, perforación intestinal inminente o estenosis intestinal, los pacientes deben someterse a una intervención quirúrgica de urgencia.
Mensajes para llevarse a casa
- En todas las vasculitis, el primer objetivo del tratamiento es lograr una remisión clínica lo más completa y rápida posible. A ésta le sigue la fase de mantenimiento de la remisión, que puede durar varios años.
- El MTX no debe combinarse nunca con sulfometoxazol-trimetoprima en el tratamiento de la RZA, ya que de lo contrario podría producirse una mielosupresión grave.
- Un nuevo y prometedor mecanismo de acción para el tratamiento de la GPA generalizada, así como de otras AAV, es la inhibición del sistema del complemento.
- La vasculitis sistémica de pequeños vasos en la GPA puede afectar a todos los órganos. La infección de los riñones suele dar lugar a una glomerulonefritis “pauciinmune” (RPGN) rápidamente progresiva y necrotizante, que puede desembocar en una insuficiencia renal grave si no se trata.
Literatura:
- Jardel S, et al: Mortalidad en las vasculitis necrotizantes sistémicas: Un análisis retrospectivo del registro del Grupo francés de estudio de la vasculitis. Autoinmune Rev 2018 Jul; 17(7): 653-659.
- Jennette JC, Falk RJ, Bacon PA, et al: 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013 Jan; 65(1): 1-11.
- Chung L, et al: Uso satisfactorio del rituximab para la vasculitis cutánea. Arch Dermatol 2006; 142(11): 1407-1410; doi:10.1001/archderm.142.11.1407.
- www.rheuma-net.ch/de/fachinformationen/behandlungsempfehlungen
- Villiger PM, Adler S, Kuchen S, et al: Tocilizumab para la inducción y el mantenimiento de la remisión en la arteritis de células gigantes: un ensayo de fase 2, aleatorizado, doble ciego y controlado con placebo. Lancet 2016; 387: 1921-1927.
- Stone JH, Tuckwell K, Dimonaco S et al: Ensayo de tocilizumab en la arteritis de células gigantes. N Engl J Med 2017; 377: 317-328.
- Christ L, et al: A Proof of Concept Study to Assess the Efficacy of Tocilizumab in Combination with Ultra-Short Glucocorticoid Administration to Treat Newly Diagnosed Giant Cell Arteritis – a 24 Week Analysis. Artritis Reumatol 2020; 72 (suppl 10).
- Adler S, et al: Riesgo de recaída tras la interrupción del tratamiento con tocilizumab en la arteritis de células gigantes. Reumatología (Oxford) 2019 Sep 1; 58(9): 1639-1643.
- Hellmich B, et al.: Actualización 2018 de las recomendaciones EULAR para el tratamiento de la vasculitis de grandes vasos. Ann Rheum Dis 2020 Jan; 79(1): 19-30.
- Gadola SD, Gross WL: El renacimiento de la inflamación granulomatosa en la AAV. Nat Rev Rheumatol 2012 Ene 10; 8(2): 74-76.
- Holle JU, et al: Las masas orbitarias en la granulomatosis con poliangeítis se asocian a un curso refractario y a una elevada carga de daño local. Reumatología 2013; 52: 875882.
- DeRemee RA, et al: Granulomatosis de Wegener: observaciones sobre el tratamiento con agentes antimicrobianos. Informes de casos Mayo Clin Proc 1985 Ene; 60(1): 27-32; doi: 10.1016/s0025-6196(12)65279-3.
- Stegeman CA, et al: Asociación del transporte nasal crónico de Staphylococcus aureus y mayores tasas de recaída en la granulomatosis de Wegener. Ann Intern Med 1994 Ene 1; 120(1): 12-17.
- Rennie Rhee D, et al: Bacterias nasales asociadas con la actividad de la enfermedad y los niveles de ANCA en la granulomatosis con poliangeítis. Artritis Reumatol 2020; 72 (suppl 10).
- Taylor GB, et al: Una revisión clinicopatológica de 34 casos de enfermedad inflamatoria mamaria que muestra una asociación entre la infección por corinebacterias y la mastitis granulomatosa. Patología 2003 abr; 35(2): 109-119.
- Dobinson HC, et al: Opciones de tratamiento antimicrobiano para la mastitis granulomatosa causada por especies de Corynebacterium. J Clin Microbiol 2015 Sep; 53(9): 2895-2899.
- Holle JU, Dubrau C, Herlyn K, et al: Rituximab para la granulomatosis con poliangitis refractaria (granulomatosis de Wegener): comparación de la eficacia en las manifestaciones granulomatosas frente a las vasculíticas. Ann Rheum Dis 2012; 71: 327-333.
- Voswinkel J, Mueller A, Kraemer JA, et al: Maduración de los linfocitos B en la granulomatosis de Wegener: un análisis comparativo de los genes VH de las lesiones endonasales, Ann Rheum Dis 2006; 65: 859-864.
- Voswinkel J, Assmann G, Held G et al: Single cell analysis of B lymphocytes from Wegener’s granulomatosis: B cell receptors display affinity maturation within the granulomatous lesions. Clin Exp Immunol 2008; 154: 339-345.
- Knight A, et al: Cáncer de vejiga urinaria en la granulomatosis de Wegener: riesgos y relación con la ciclofosfamida. Ann Rheum Dis 2004.
- Thiel J, et al: Cinética de repoblación de células B tras el tratamiento con rituximab en las vasculitis asociadas a ANCA en comparación con la artritis reumatoide y las enfermedades del tejido conectivo: un estudio observacional longitudinal en 120 pacientes. Arthritis Res Ther 2017 18 de mayo; 19(1): 101.
- Merkel P, et al: Efecto sobre la función renal del inhibidor del receptor del complemento C5a Avacopan en la vasculitis asociada a ANCA. Artritis Reumatol 2020; 72 (suplemento 10).
- Huizenga N, et al: Tratamiento de la vasculitis agresiva asociada a anticuerpos citoplasmáticos antineutrófilos con eculizumab. Kidney Int Rep 2020 abr; 5(4): 542-545.
- Smtih R, et al: Seguimiento ampliado de pacientes reclutados en un ensayo aleatorizado y controlado de rituximab frente a azatioprina tras la inducción de la remisión con rituximab en pacientes con vasculitis asociada a ANCA y enfermedad recidivante. Artritis Reumatol 2020; 72 (suppl 10).
- Guillevin L, et al: Rituximab frente a azatioprina para el mantenimiento en la vasculitis asociada a ANCA. N Engl J Med 2014 Nov 6; 371(19): 1771-1780.
- Tzelepis GE, et al: Prevalencia y pronóstico de la fibrosis pulmonar en la poliangeítis microscópica. Revista Respiratoria Europea 2010; 36: 116-121.
- Sinico RA, et al: Afectación renal en el síndrome de Churg-Strauss. Am J Kidney Dis 2006 mayo; 47(5): 770-779.
- Wolf J, et al: Complicaciones neurológicas del síndrome de Churg-Strauss: un estudio prospectivo monocéntrico. Neurología actual 2009; 36: V188.
- Conron M, Beynon HL: Síndrome de Churg-Strauss. Thorax 2000 Oct; 55(10): 870-877.
- Solans R, et al: Síndrome de Churg-Strauss: resultados y seguimiento a largo plazo de 32 pacientes. Reumatología (Oxford) 2001; 40: 763-771.
- Braun G, et al: Vasculitis crioglobulinémica: clasificación y aspectos clínicos y terapéuticos. Postgrad Med J 2007 feb; 83(976): 87-94; doi: 10.1136/pgmj.2006.046078.
- Bettiol A, et al: Tratamiento de los diferentes fenotipos del síndrome de Behçet. Front Immunol 2019 dic 6; 10: 2830: doi: 10.3389/fimmu.2019.02830. eCollection 2019.
InFo DOLOR Y GERIATURA 2020; 2(2): 12-19