Ci sono molte mutazioni diverse che possono essere alla base della distrofia retinica ereditaria. Grazie allo sviluppo dei moderni metodi di analisi genetica molecolare, oggi è possibile identificare la maggior parte delle cause genetiche. È disponibile una terapia genica innovativa per i pazienti con distrofia retinica dovuta a mutazioni bialleliche di RPE65.
Il termine ombrello “distrofie retiniche ereditarie” (IRD; malattie retiniche ereditarie) copre un gruppo eterogeneo di malattie con fenotipi sovrapposti, caratterizzate da degenerazione progressiva e disfunzione della retina [1]. Ciò che queste malattie hanno in comune è che colpiscono l’intera retina durante il decorso della malattia e che la visione si deteriora lentamente e progressivamente nel corso della vita di una persona. L’IRD può essere causata da mutazioni in >250 geni diversi [2]. La patogenesi molecolare, così come il quadro clinico e il decorso della malattia sono definiti in modo significativo dal tipo e dalla natura del difetto genetico [3]. A seconda del gene mutato, possono verificarsi vari disturbi dello sviluppo e funzionali della retina, come disturbi del metabolismo retinico, delle strutture cellulari o della fototrasduzione. La forma più comune di IRD è la retinite pigmentosa, ma esistono anche l’amaurosi congenita di Leber e la distrofia retinica ad esordio precoce (EORD) [2].
La diagnosi precoce è fondamentale
Nella retinite pigmentosa, la degenerazione dei bastoncelli si verifica nelle prime fasi della malattia, con conseguente cecità notturna. I sintomi caratteristici dell’amaurosi congenita di Leber comprendono una drastica riduzione dell’acuità visiva e difetti del campo visivo. La diagnosi corretta e precoce della malattia è molto importante, così come una descrizione morfologica e funzionale dettagliata della condizione attuale della retina, a seconda delle mutazioni sottostanti nel gene RPE65. Questo perché la progressione rapida e grave della malattia rende necessario un intervento precoce, finché le cellule dell’epitelio pigmentato retinico come cellule bersaglio e anche i fotorecettori sono ancora presenti. Nell’ambito della diagnostica funzionale oftalmologica e soprattutto della diagnostica per immagini non invasiva (ad esempio, tomografia a coerenza ottica, autofluorescenza del fundus o del vicino infrarosso), è possibile la descrizione clinica del fenotipo [5,6]. Per identificare la causa genetica delle distrofie retiniche ereditarie, è indispensabile l’uso di metodi genetici molecolari come il sequenziamento di nuova generazione.
L’imaging ad autofluorescenza fornisce risultati importanti
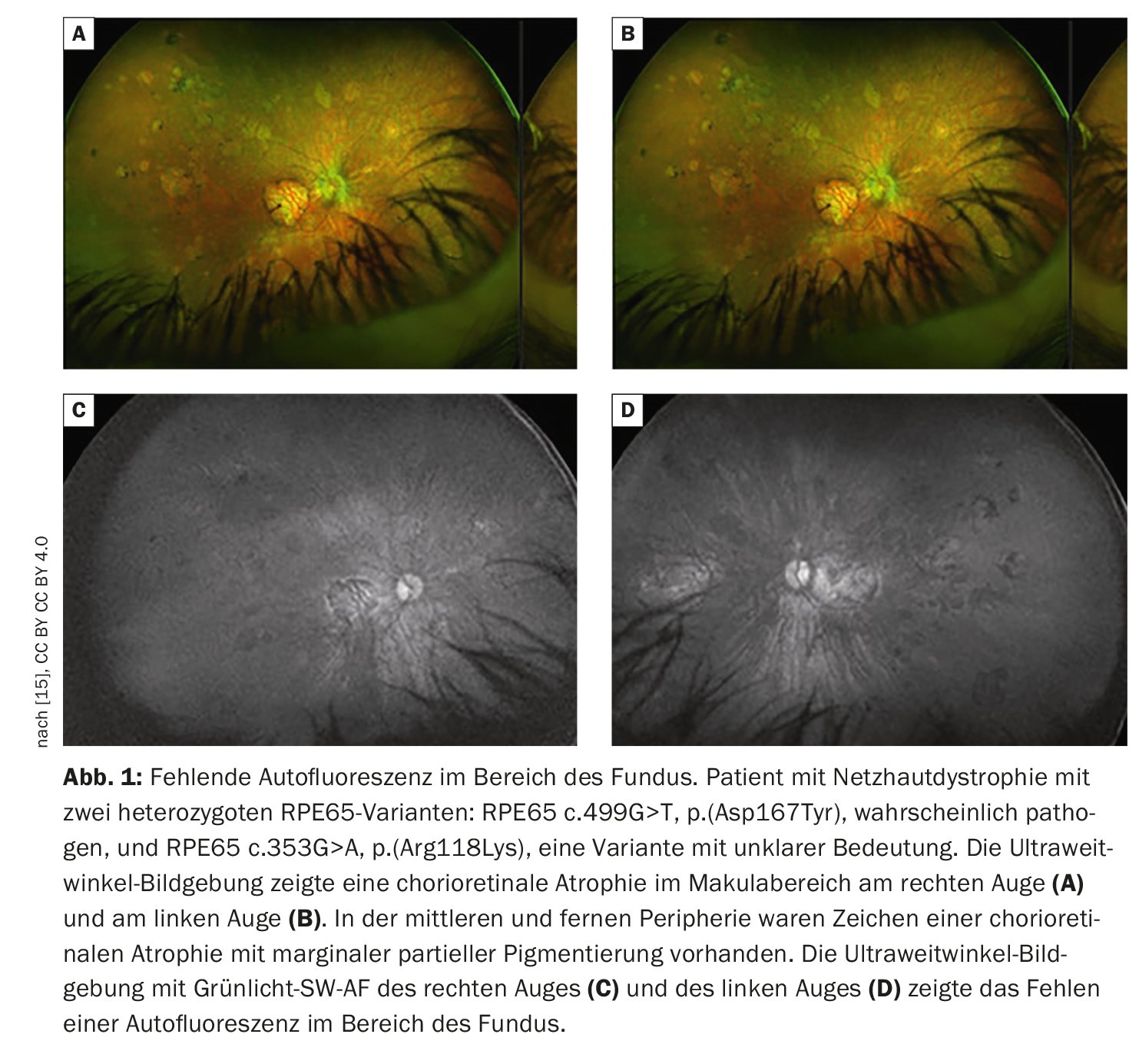
L’imaging retinico fornisce una visione più dettagliata della patologia retinica rispetto all’esame funduscopico. Oltre alla fotografia convenzionale del fondo a colori, la tomografia a coerenza ottica (OCT) ad alta risoluzione e l’autofluorescenza del fondo, che può visualizzare i fluorofori del fondo oculare per mezzo della luce a onde corte, sono i metodi più consolidati. L’autofluorescenza del fondo mostra la distribuzione dei fluorofori della parte posteriore dell’occhio, principalmente utilizzando la luce di eccitazione a onde corte nella gamma del blu o del verde. Nei pazienti con mutazioni nel gene RPE65, l’autofluorescenza assente o almeno ridotta nella regione del fondo (Fig. 1) è una caratteristica clinicamente significativa dovuta al metabolismo difettoso dei retinoidi nei fotorecettori e nelle cellule RPE [9]. Per esempio, nella retinite pigmentosa c’è tipicamente un anello concentrico di aumento dell’autofluorescenza per il quale non c’è una correlazione visibile a livello funduscopico [10–12]. Sebbene l’origine esatta di questo fenomeno sia incompleta, gli studi OCT hanno dimostrato che l’anello corrisponde alla perdita della banda ellissoidale e a un grave assottigliamento o addirittura alla perdita dello strato dei fotorecettori [10].
Test genetici molecolari per il rilevamento della causa genetica
I fenotipi nelle varianti di geni diversi spesso si sovrappongono e una notevole variabilità è possibile nelle varianti di un gene. Pertanto, i test genetici molecolari sono essenziali e costituiscono una base importante per la selezione dell’opzione terapeutica appropriata [6]. Nel 55-80% dei casi, la causa genetica delle distrofie retiniche ereditarie può essere identificata mediante test genetici molecolari [4]. Nei pazienti con mutazioni bialleliche nel gene RPE65, l’isomerasi RPE65 non è espressa affatto o è espressa come proteina non funzionale nell’epitelio pigmentato retinico (RPE). Per definizione, per la diagnosi di distrofia retinica associata alla mutazione biallelica RPE65, entrambe le copie del gene RPE65 devono essere presenti come una o due varianti patogene distinte [8]. Il fenotipo clinico in cui si manifestano le mutazioni bialleliche di RPE65 è solitamente associato all’amaurosi congenita di Leber o alla retinite pigmentosa. La mancanza di un’isomerasi RPE65 funzionale significa che l’all-trans-retinale prodotto nella cascata di fototrasduzione non viene convertito in 11-cis-retinale, il che ostacola la rigenerazione del pigmento visivo rodopsina [1]. Questo disturbo del metabolismo retinico fa sì che i fotorecettori perdano la capacità di rispondere agli stimoli luminosi. Inoltre, l’accumulo di intermedi tossici porta alla morte delle cellule epiteliali del pigmento retinico, con conseguente ulteriore disfunzione dei fotorecettori. Questo effetto è più pronunciato nei bastoncelli fotorecettori rispetto ai coni fotorecettori, poiché questi ultimi hanno ancora un percorso metabolico alternativo per la rigenerazione del pigmento visivo attraverso le cosiddette cellule di Müller [1].
Il farmaco per la terapia genica come opzione di trattamento per la mutazione del gene RPE65
L’idea di base è che, aggiungendo una copia corretta mediante la terapia genica retinica, si possa ripristinare la funzione della retina. I vettori virali ricombinanti adeno-associati (AAV) sono molto adatti per effettuare il trasferimento di geni, in quanto consentono un’espressione duratura e specifica del tipo di cellula nella retina [9]. Voretigen Neparvovec è un vettore di trasferimento genico che utilizza il capside di un vettore virale adeno-associato di sierotipo 2 (AAV2) come veicolo di trasporto per il cDNA della proteina 65 kDa specifica dell’epitelio pigmentato retinico umano (hRPE65) nella retina [13].
| Neparvovec pre-etigenico con mutazione biallelica RPE65 L’efficacia di Luxturna® (voretigen neparvovec) è stata studiata in un totale di 41 pazienti, tutti con distrofia retinica ereditaria [13]. Nello studio principale, hanno partecipato 11 adulti (36%) e 20 bambini di età superiore ai 4 anni (64%). L’età media era di 15 anni. Per dimostrare l’efficacia di Luxturna® nello studio principale, è stata misurata la visione funzionale. Si tratta di acuità visiva, campo visivo e percezione e/o visione in condizioni di scarsa illuminazione. Dopo un anno di trattamento, i pazienti nel braccio di trattamento Luxturna® sono stati in grado di completare un corso in modo più preciso, più veloce e in condizioni di scarsa luminosità. Il miglioramento della visione funzionale è stato mantenuto per un periodo di almeno tre anni. È in corso un follow-up di 15 anni dei 41 partecipanti allo studio. |
Luxturna® è un farmaco di terapia genica contenente il principio attivo voretigen neparvovec ed è stato approvato in Svizzera dal febbraio 2020 per il trattamento di adulti e bambini con perdita della vista dovuta alla distrofia retinica ereditaria con mutazione biallelica RPE65 (box) [13]. Il trattamento con Luxturna® richiede che la mutazione RPE65 sia stata confermata da un test genetico e che ci siano ancora sufficienti cellule funzionali nella retina del paziente [13]. Luxturna® viene applicato per via microchirurgica sotto la retina [13,14]. In Svizzera, un centro esclusivo per questa terapia genica si trova presso l’Ospedale Universitario di Basilea (USB) [14]. Il Prof. Dr. med. Hendrik Scholl è capo e primario della Clinica oculistica dell’USB, il Prof. Dr. med. Christian Prünte è primario clinico e si è specializzato, tra l’altro, in microchirurgia. Nel novembre 2022, l’USB ha annunciato che un team di dodici membri guidato dal Prof. Scholl e dal Prof. Prünte ha eseguito il primo intervento microchirurgico in Svizzera per l’applicazione di Luxturna®. Il paziente di 51 anni affetto da distrofia retinica ereditaria è stato sottoposto al trattamento del secondo occhio nel marzo 2022, dopo l’intervento chirurgico sull’altro occhio effettuato un anno prima [14]. Entrambi gli interventi si sono svolti in modo ottimale.
Messaggi da portare a casa
- Ad oggi, sono note più di 250 mutazioni alla base della distrofia retinica ereditaria. Nel 55-80% dei casi, la causa genetica può essere identificata con un test genetico molecolare [4].
- Il difetto genetico sottostante definisce la patogenesi molecolare, il quadro clinico e il decorso della malattia. Le varianti del gene RPE65 sono la causa dello 0,6-6% di tutti i casi di retinite pigmentosa e del 3-16% dei casi di amaurosi congenita di Leber/distrofia retinica ad insorgenza precoce [2].
- Nell’imaging retinico, le mutazioni del gene RPE65 sono caratterizzate da un’autofluorescenza assente o ridotta nel fondo.
- Luxturna® (voretigene neparvovec) è la prima terapia genica disponibile per il trattamento della distrofia retinica ereditaria con mutazione biallelica RPE65 [13].
Letteratura:
- Dias MF, et al: Genetica molecolare e terapie emergenti per la retinite pigmentosa: ricerca di base e prospettive cliniche. Progress in Retinal and Eye Research 2017 (63): 107-131.
- Aoun M, et al: Distrofie retiniche ereditarie dovute a varianti di RPE65: Dalla diagnosi genetica alla terapia. Compass Ophthalmol 2021; 7: 115-123.
- Nash BM, et al: Distrofie retiniche, applicazioni genomiche nella diagnosi e prospettive di terapia. Pediatria Traslazionale 2015; 4 (2): 139-163.
- Kohl S, Biskup S: Test diagnostici genetici nelle distrofie retiniche ereditarie. Clin Monbl Ophthalmology 2013; 230(3): 243-246.
- Bolz HJ: Analisi diagnostiche dei geni della distrofia retinica: stato attuale e prospettive. Clin Monbl Ophthalmology 2021; 238: 261-266.
- Kellner U, et al.: Diagnostica delle distrofie retiniche ereditarie. Importanza della diagnostica genetica molecolare dal punto di vista del paziente. Oftalmologia 2022; 119: 820-826.
- Dockery A, et al: Applicazioni del sequenziamento di nuova generazione per le malattie ereditarie della retina. Int J Mol Sci 2021; 22: 5684.
- Blue Cross and Blue Shield of Kansas City (BCBSKC) 2018. Terapia genica per la distrofia retinica ereditaria: Politica numero 2.04.144.
- Stieger K, Lorenz B: Terapia genica per le malattie degenerative della retina, www.kaden-verlag.de/fileadmin/images/ZPA_direkt/CME_ZPA_12-09.pdf,(ultimo accesso 04.04.2023)
- Birtel J, et al: Diagnosi delle malattie retiniche ereditarie Klin Monatsbl Augenheilkd 2021; 238: 249-260.
- Robson AG, et al: Imaging seriale e correlazioni struttura-funzione degli anelli ad alta densità di autofluorescenza del fondo nella retinite pigmentosa. Retina 2011; 31: 1670-1679.
- Lima LH, et al: Costrizione progressiva dell’anello iperautofluorescente nella retinite pigmentosa. Am J Ophthalmol 2012; 153: 718-727.
- Informazioni sui farmaci, www.swissmedicinfo.ch,(ultimo accesso 04.04.2023)
- “Primo utilizzo della terapia genica contro la cecità in Svizzera”, www.unispital-basel.ch/newscenter/zuweisenden-blog/15-07-2022,(ultimo accesso 04.04.2023)
- Bjeloš M, et al.: RPE65 c.353G>A, p.(Arg118Lys): Una nuova mutazione puntiforme associata alla retinite pigmentosa e all’atrofia maculare. Giornale Internazionale di Scienze Molecolari. 2022; 23(18):10513. www.mdpi.com/1422-0067/23/18/10513,(ultimo accesso 04.04.2023).
PRATICA GP 2023; 18(4): 46-47