A cardiomiopatia hipertrófica (CMH) é a cardiomiopatia genética mais comum em todo o mundo e afecta cerca de 1 em cada 500 pessoas. As intervenções terapêuticas actuais incluem a otimização do estilo de vida, a medicação, as terapias de redução do septo e, raramente, o transplante cardíaco. Os avanços na compreensão das variantes genéticas causadoras de doença na CMH e dos seus mecanismos moleculares abriram o potencial para terapias direccionadas e para a implementação da medicina de precisão e personalizada. Os resultados da investigação pré-clínica são promissores e levantam a questão de saber se, no futuro, será possível uma cura para alguns subtipos de MCH.
A cardiomiopatia hipertrófica (CMH) é uma doença miocárdica primária caracterizada por hipertrofia ventricular esquerda inexplicada que ocorre na ausência de condições de stress anormais, como a hipertensão ou a estenose aórtica. A apresentação clínica da CMH é muito variável, desde indivíduos assintomáticos até aos que apresentam sintomas graves, como insuficiência cardíaca, arritmias e morte súbita cardíaca (MSC). A base genética da CMH é também heterogénea, com mutações em pelo menos oito genes do sarcómero identificadas como patogénicas e uma proporção significativa de casos associados a variantes genéticas de significado indeterminado (VUS).
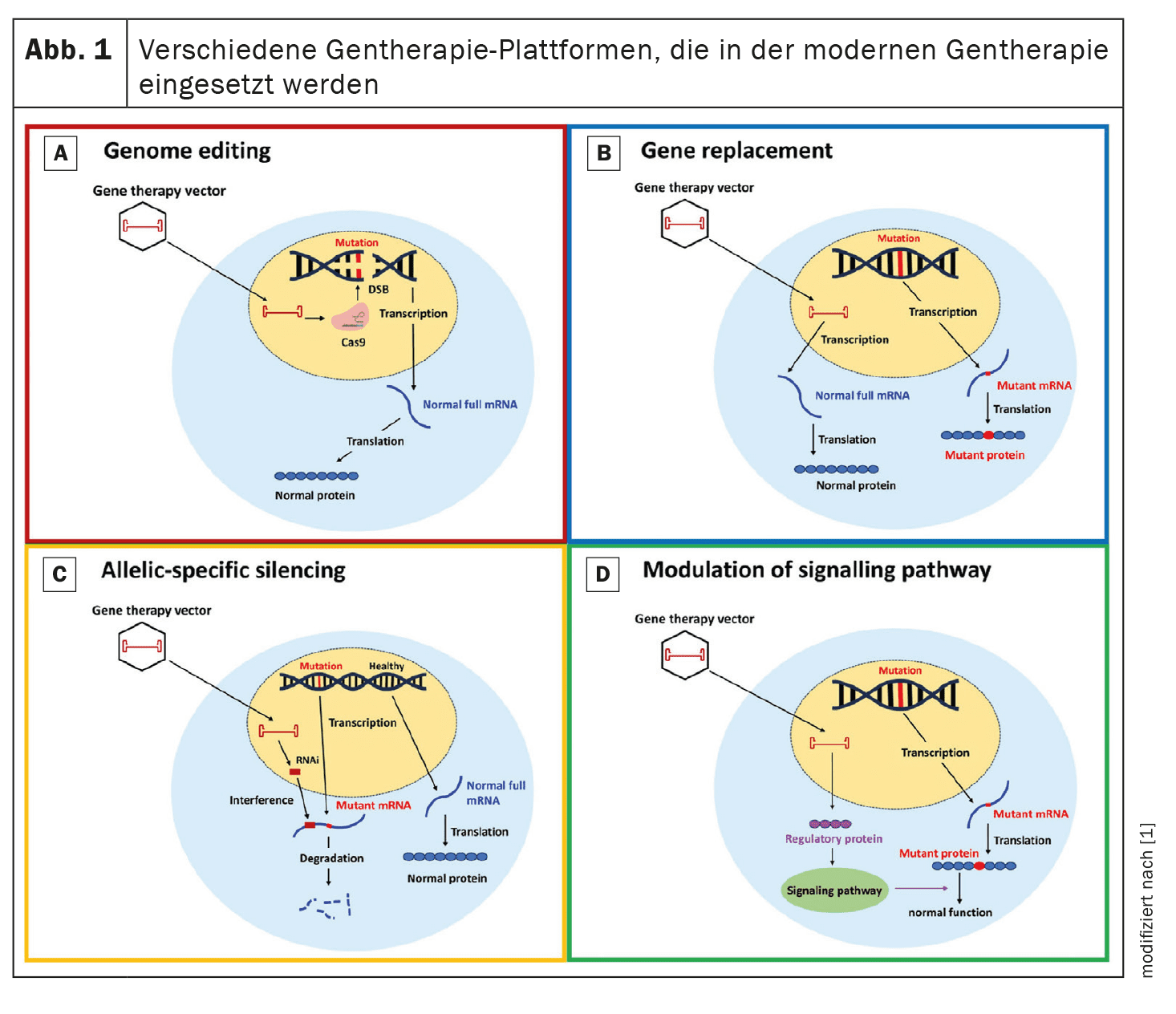
Panorama terapêutico atual para a CMH
Medidas relativas ao estilo de vida: A otimização do estilo de vida é crucial para a gestão da MCH. Evitar a hipertensão não controlada e a obesidade é essencial para prevenir a exacerbação do fenótipo da MCH. Estudos recentes também sublinharam os benefícios do exercício regular, ligeiro a moderado, na melhoria da qualidade de vida e dos resultados cardiovasculares nos doentes com MCH. Anteriormente, os doentes com MCH eram desaconselhados a praticar atividade física intensa, mas estudos recentes sugerem que o exercício moderado pode ser seguro e benéfico.
Terapia medicamentosa: Medicamentos como os beta-bloqueadores e os bloqueadores dos canais de cálcio são frequentemente utilizados para aliviar os sintomas e reduzir o risco de complicações da CMH. Estes medicamentos actuam diminuindo a frequência cardíaca e reduzindo a contratilidade do coração, o que ajuda a reduzir a obstrução da via de saída do ventrículo esquerdo. Terapias mais recentes, como o mavacamten, um inibidor da miosina, mostraram resultados promissores em estudos recentes, melhorando os sintomas subjectivos e objectivos em doentes com MCH obstrutiva. O mavacamten modifica diretamente as cadeias pesadas da β-miosina para reduzir a afinidade entre a actina e a miosina e, assim, normalizar a função cardíaca.
Terapias de intervenção: Os procedimentos de intervenção, incluindo a miectomia septal e a ablação septal com álcool, são realizados para aliviar a obstrução da via de saída do ventrículo esquerdo (VSVE). A miectomia septal é uma intervenção cirúrgica em que uma parte do septo espessado é removida para melhorar o fluxo sanguíneo do ventrículo esquerdo. Este método é normalmente utilizado em doentes mais jovens. A ablação septal com álcool, uma alternativa menos invasiva, é mais comummente realizada em doentes mais velhos. Envolve a injeção de álcool nas artérias septais para provocar um enfarte controlado e reduzir a hipertrofia. Apesar das taxas mais elevadas de arritmias e cicatrizes em comparação com a miectomia septal, a ablação septal com álcool pode alcançar excelentes resultados em centros experientes.
Prevenção da morte súbita cardíaca: Os cardioversores-desfibrilhadores implantáveis (CDI) são recomendados para pessoas com elevado risco de morte súbita cardíaca . Os avanços na estratificação do risco e a disponibilidade de CDIs reduziram significativamente a mortalidade relacionada com a MCH. A avaliação do risco é realizada utilizando calculadoras de risco baseadas em directrizes que têm em conta factores como a espessura do septo, a presença de taquicardia ventricular não sustentada e a história familiar.
Transplante de coração: Em casos raros, quando os doentes não respondem a todos os outros tratamentos e desenvolvem insuficiência cardíaca avançada, pode ser necessário um transplante de coração. Estes doentes constituem cerca de 1,6% dos que aguardam um transplante cardíaco nos EUA. Apesar da sua raridade, a taxa de sobrevivência após transplante cardíaco em doentes com CMH é semelhante à de doentes com outras cardiomiopatias.
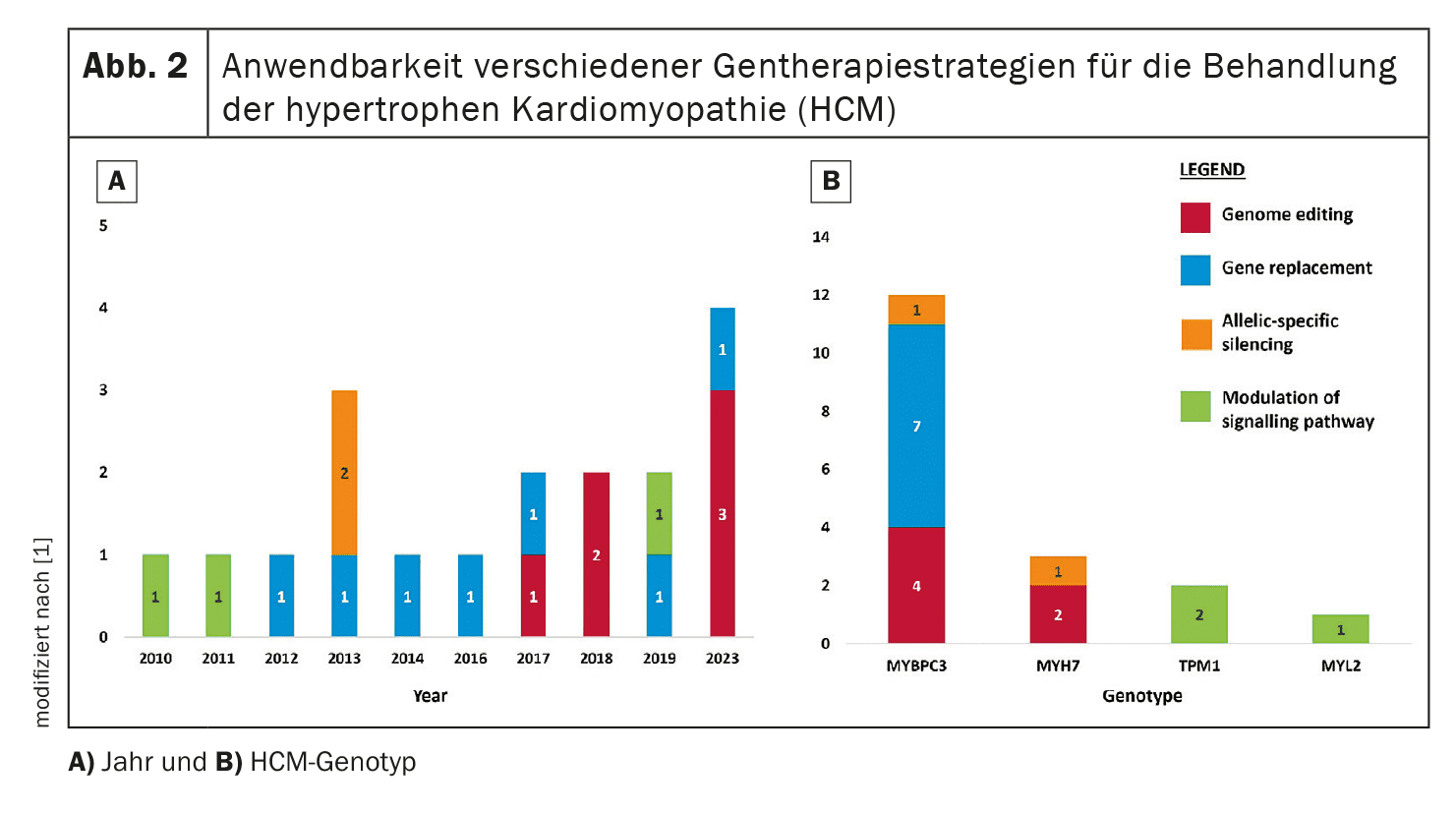
Terapia genética – a nova fronteira?
A terapia genética tem como objetivo corrigir ou atenuar as mutações genéticas responsáveis pela CMH. Nesta revisão, são analisadas quatro abordagens principais: Edição do genoma, substituição de genes, silenciamento específico de alelos e modulação de vias.
Edição do genoma: A edição do genoma com a tecnologia CRISPR/Cas9 demonstrou o potencial para corrigir mutações genéticas associadas à CMH em modelos pré-clínicos.
A CRISPR/Cas9 utiliza uma nuclease programável que gera quebras de cadeia dupla de ADN que podem ser reparadas por junção de extremidades não homólogas (NHEJ) ou recombinação homóloga (HDR).
No entanto, desafios como os efeitos fora do alvo e a necessidade de métodos de entrega precisos continuam a ser obstáculos significativos.
A investigação centra-se na melhoria da especificidade e da eficiência desta tecnologia para minimizar as alterações genéticas não intencionais. Substituição de genes: A terapia de substituição de genes envolve a introdução de uma cópia funcional do gene mutado.
Esta abordagem é particularmente relevante para as mutações que conduzem à haploinsuficiência.
Estudos em curso, como a utilização de vectores de vírus adeno-associados (AAV), pretendem avaliar a eficácia e segurança desta estratégia em doentes com MCH.
Por exemplo, a substituição do gene MYBPC3, que está frequentemente mutado na MCH, está atualmente a ser investigada em ensaios clínicos.
Em estudos pré-clínicos, a substituição do gene defeituoso demonstrou melhorar a função miocárdica e prevenir os fenótipos da CMH.
Silenciamento específico do alelo: O silenciamento específico do alelo tem como objetivo suprimir o alelo mutante, preservando a função do alelo normal. Esta abordagem utiliza pequenos RNAs de interferência (siRNAs) para degradar seletivamente o mRNA mutante e reduzir a produção de proteínas anormais. Este método é particularmente útil para mutações dominantes em que a proteína mutante tem um efeito deletério. Estudos pré-clínicos demonstraram que os siRNAs podem reduzir eficazmente a expressão do alelo mutante, resultando numa melhoria da função cardíaca e numa redução da hipertrofia.
Modulação das vias de sinalização: A modulação das principais vias de sinalização envolvidas na patogénese da MCH oferece outra abordagem terapêutica. Por exemplo, o aumento da expressão de SERCA2a, uma proteína envolvida no equilíbrio do cálcio, demonstrou potencial em modelos pré-clínicos para reduzir a hipertrofia e melhorar a função cardíaca. Outra abordagem envolve a modulação da fosforilação da cadeia leve reguladora da miosina (miosina RLC) para melhorar a função contrátil cardíaca.
Desafios e direcções futuras
Apesar dos progressos promissores, existem vários desafios que impedem a tradução clínica das terapias genéticas para a CMH. Estes incluem:
- Preocupações de segurança: Os riscos associados aos vectores AAV, como as respostas imunitárias e os efeitos fora do alvo, têm de ser abordados de forma exaustiva. As mortes recentes associadas às terapias genéticas baseadas em AAV recordam os perigos potenciais e a necessidade de melhorar ainda mais a segurança destas tecnologias.
- Métodos de administração: A administração eficiente e direccionada de terapias genéticas ao tecido cardíaco continua a ser um desafio técnico. O desenvolvimento de vectores com tropismo cardíaco específico e melhorado poderia ajudar a minimizar as transduções fora do alvo e a reduzir as doses necessárias.
- Heterogeneidade genética: A diversidade genética da MCH dificulta o desenvolvimento de abordagens universais de terapia genética. Como a CMH é causada por um grande número de mutações, o desenvolvimento de gRNAs específicos para cada mutação e a limitação por sequências PAM requerem mais investigação e otimização.
- Ética e acesso equitativo: É fundamental garantir que as terapias genéticas sejam acessíveis a todos os doentes, independentemente do seu estatuto socioeconómico. Deve ser dada prioridade à distribuição equitativa destes tratamentos avançados para evitar desigualdades na saúde.
Conclusão
Os avanços na terapia genética são muito promissores para o futuro tratamento da CMH (HCM). Embora a investigação pré-clínica tenha demonstrado potencial, a transposição destas terapias para a prática clínica exigirá desafios científicos, técnicos e éticos significativos. Deve ser assegurado um acesso equitativo a estes tratamentos avançados, para que se concretize todo o seu potencial de melhoria dos resultados para os doentes com MCH. A continuação da investigação e do desenvolvimento de terapias genéticas seguras e eficazes poderá revolucionar a forma como a MCH e outras cardiomiopatias genéticas são tratadas.
Fonte:
- Paratz ED, Mundisugih J, Rowe SJ, et al.: Gene Therapy in Cardiology: Is a Cure for Hypertrophic Cardiomyopathy on the Horizon? Can J Cardiol 2024 May; 40(5): 777–788. doi: 10.1016/j.cjca.2023.11.024. Epub 2023 Nov 25. PMID: 38013066.
CARDIOVASC 2024; 23(2): 24–25