If a catecholamine-secreting tumor is suspected, determination of metanephrines in free plasma is a sensitive diagnostic method. Paroxysmal hypertension is a common symptom. Depending on the laboratory findings, an imaging examination should be performed for tumor localization. For non-resectable or metastatic tumors, there are new experimental therapies based on the individual genetic and molecular signature in which much hope is placed. In this context, genetic analyses are gaining importance.
Pheochromocytomas (PCCs) are catecholamine-producing tumors that arise from the chromaffin cells of the adrenal medulla. Paragangliomas (PGLs) are noradrenaline- and dopamine-producing tumors of the sympathetic and parasympathetic paraganglia [1]. The clinical presentation and initial diagnostic approach are very similar in both catecholamine-secreting tumor types [2]. A typical symptom of pheochromocytoma is hypertension, which occurs paroxysmally in about half of affected patients. Other signs of the disease include, for example, sweating, palpitations, headache, tremor or nausea. In most cases, PGL are readily curable by complete resection, but there is considerable variation in malignancy risk depending on genotype and tumor location. Tumors that metastasize are considered malignant, which is approximately 10-15% of all PCCs and 35-40% of all PGLs [1]. Genetic testing should be performed in all patients with pheochromocytomas/paragangliomas (PPGL), emphasized Prof. Svenja Nölting, MD, senior physician at the Department of Endocrinology, Diabetology and Clinical Nutrition, University Hospital Zurich, at the annual meeting of the Swiss Society of Endocrinology and Diabetology (SGED) last November [1].

Cluster typing: genetic and molecular signature
“About 70% of all PPGLs can be assigned to one of three molecular clusters based on their underlying mutation,” the speaker explained [1,3]. Most tumors belong to clusters 1 and 2; little is known about cluster 3 so far. Cluster 1 tumors are based on activation of pseudohypoxia signaling pathways, and cluster 2 tumors are based on activation of tyrosine kinase-dependent signaling pathways [4]. Autosomal dominant germline mutations are present in 30-35% of cases, and 35-40% are due to somatic mutations. Cluster 1 tumors are more often extraadrenal localized and have the highest risk of metastasis, while cluster 2 tumors are mostly adrenal localized and have a low risk of metastasis [4].
Diagnostics: Metanephrines in free plasma are meaningful markers
Patients with known germline mutation, but also with clinical suspicion of pheochromocytoma/paraganglioma (PPGL) or if a history of adrenal incidentaloma or PPGL has been reported, should be screened [4]. As a first step, the following biochemical laboratory parameters should be collected [1]:
- Metanephrines in free plasma (=metabolites of adrenaline)
- Normetanephrines (=metabolites of noradrenaline)
- 3-methoxytyramine (=metabolites of dopamine)
It is recommended that this be collected by mass spectrometry, as this can achieve better sensitivity than other methods. Prior to blood collection, patients should refrain from nicotine and coffee, and from taking SSRIs and/or tricyclic antidepressants, crystal meth, or cocaine, among other medications. In addition to the parameters mentioned, it is also useful to determine chromogranin A, as this is a good marker, especially for asymptomatic tumors. In this case, the patient should refrain from taking proton pump inhibitors (PPI) one week before the examination. Catecholine determination alone is not useful, she said, as this could lead to a false-negative finding. If the determined metanephrine values are above twice the normal value, imaging is indicated in a further clarification step [1]. Initially, MRI/CT of the abdomen and pelvic region is recommended; if the findings are negative or if an extra-adrenal tumor/PGL is already present, functional imaging is required. If the size of the adrenal tumor is <5 cm, Prof. Nölting advises the use of DOPA PET/CT. DOTA-SSA-PET/CT should be performed in the presence of extra-adrenal tumors and negative anatomic imaging findings or when metastases are already present, or the size of the adrenal tumor is >5 cm, the speaker explained [1].
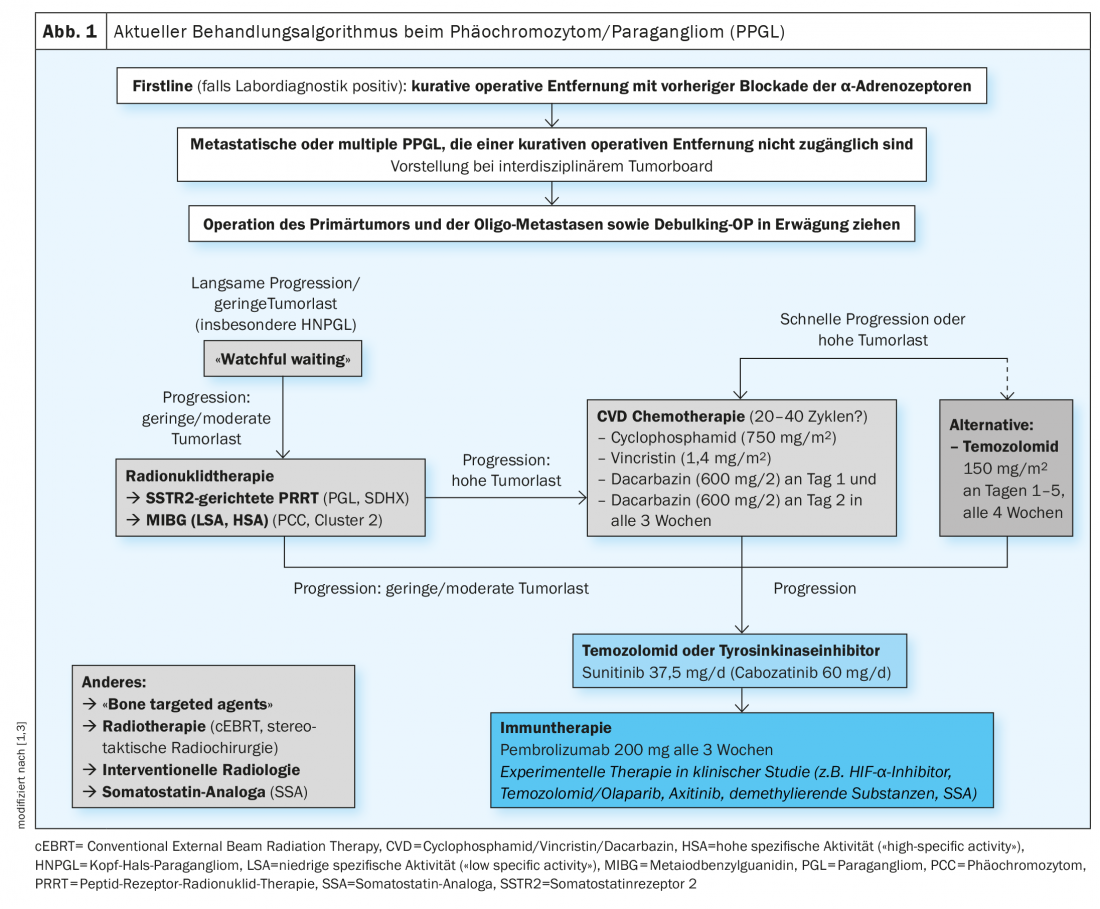
Individualized treatment strategy: new experimental therapies.
The current treatment algorithm is shown in Figure 1 [1,3]. Surgical excision is considered the first-line therapy of localized PPGL. This can usually be done minimally invasively and should be performed at an experienced center [4]. To avoid complications, preoperative drug blockade of α-adrenoceptors is performed. For metastatic PPGL, treatment is currently with radionuclide therapy, chemotherapy, or tyrosine kinase inhibitors, although there are no officially approved therapies yet. Other novel treatment strategies are currently under clinical investigation. So far, cluster-specific therapy of inoperable or metastatic disease has not been established in clinical practice. However, this would be useful to be able to offer personalized, genetically guided treatment, according to Prof. Nölting [3]. A scientific publication has appeared in Endocrine Reviews 2021 detailing how a coherent individualized patient management plan can be developed in patients with pheochromocytoma/paraganglioma based on genetic analysis and cluster typing, respectively, to optimally tailor treatment options [3].
Congress: Swiss Society for Endocrinology and Diabetology 11.11.2021
Literature:
- Nölting S: “Genetic testing in patients with pheochromocytomas/paragangliomas”, Prof. Dr. med. Svenja Nölting, SGED 11.11.2021
- Zulewski H, Grouzmann E: Diagnosis and treatment: “Pheochromocytoma,” Swiss Med Forum 2017; 17(37): 790-796, https://medicalforum.ch/de/detail/doi/smf.2017.03057
- Nölting S, et al: Personalized management of pheochromocytoma and paraganglioma. Endocr Rev 2021 Jun 19:bnab019
- Pheochromocytoma – model disease for personalized medicine, Dtsch Med Wochenschr 2021; 146(23): 1520-1526.
- German Society for Endocrinology, Hormones and Metabolism: PROSPHEO, www.endokrinologie.net/sektion-nebenniere-steroide-hypertonie-7.php (last accessed Dec. 07, 2021).
HAUSARZT PRAXIS 2022; 17(1): 42-43
InFo ONCOLOGY & HEMATOLOGY 2022; 10(3): 18-19.