Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired disease characterized by intravascular hemolysis and hemoglobinuria. Due to a prolonged loss of hemoglobin through the urine, PNH can lead to iron deficiency and increase the suspicion of anemia. However, PNH is an extremely heterogeneous disease with diverse manifestations and often overlaps with other bone marrow diseases.
PNH is a very rare disease with an incidence of about 1.3/1,000,000 and a prevalence of about 16 per million population. The symptoms were first described as paroxysmal hemoglobinuria in 1882 by Dr. Strübing from the medical polyclinic in Greifswald. Marchiafava and Micheli introduced the term Marchiafava-Micheli sydrome, which is still used today as a synonym for the disorder. The name PNH was coined by the three characteristics of this disease: paroxysmal= occurs in episodes; nocturnal= occurs at night; hemoglobinuria= hemoglobin in urine. Nowadays, it is known that hemolysis can occur without symptoms and is constant throughout the day. In addition, only one in four patients has hemoglobin in the urine.
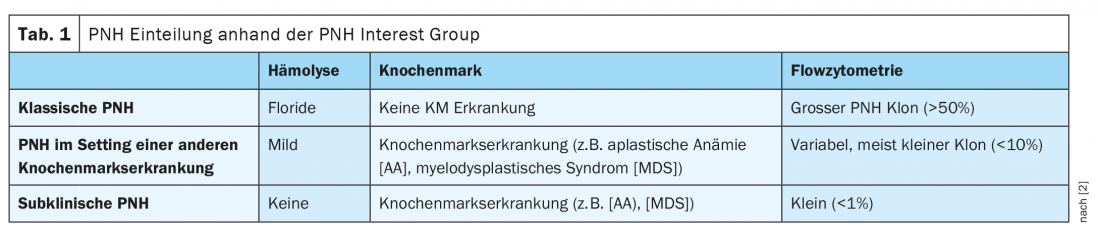
Can be divided into three forms
PNH can be divided into three forms according to the extent of hemolysis, bone marrow findings, and flow cytometry. In classic PNH, the patient has very marked hemolysis but no other bone marrow disease. In addition, these patients have a large PNH clone >50%. In addition to classic PNH, the disease can also occur in the setting of other bone marrow disorders, classically aplastic anemia. These patients are usually asymptomatic and have only mild hemolysis and a variable small PNH clone <10%. Subclinical PNH often occurs in bone marrow disorders, such as aplastic anemia or myelodysplastic syndrome. Usually, only a very small PNH clone is detected in these cases <1%, without any hemolysis signs (Table 1) [2]. In addition, PNH can overlap with other diseases at any time. Transition to aplastic anemia (AA) as well as acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) is possible.
It always starts with PIGA
PNH is due to an x-linked mutation of the gene for phosphatidyl inositol glycan A (PIGA). This enzyme is involved in the biosynthesis of the so-called GPI anchor. The GPI anchor fixes certain proteins to the surface of the cell membrane. In particular, these bound proteins include Complement Decay Accelerating Factor (CD55) and Membrane Inhibitor of Reactive Lysis (CD59). These two components serve to defend against the complimentary attack on the erythrocyte membrane. Partial or complete absence of the GPI anchor and a corresponding omission of the complement-regulating surface proteins leaves the erythrocyte membrane inadequately protected. The result is intravascular hemolysis. Among other things, intravascular hemolysis leads to the formation of free hemoglobin in the blood, which binds nitric oxide. This results in endothelial dysfunction with consecutive smooth muscle contraction and platelet activation and aggregation, leading to an increased risk of thromboembolism.
It is worth mentioning that patients with a PIGA mutation usually also have other somatic mutations. In hematology, this is then referred to as clonal changes or clones. This is a reason for different courses, and in some cases a malignant disease may develop.
Classic Triassic
Patients with PNH present with a classic triad consisting of hemolysis, thromboembolism, and possible bone marrow failure. This leads to the fact that different complaints can occur at different times. These included dysphagia (41%), pulmonary hypertension (47%), dyspnea (66%), abdominal pain (59%), renal insufficiency (64%), erectile dysfunction (47%), hemoglobinuria (26%), and thrombosis (40%). However, the main symptoms include anemia (88%) and marked fatigue (97%). In addition to these symptoms, however, atypical presentations may also occur, as a case report from Switzerland has shown. In this particular case, PNH was finally detected in a patient with myocardial infarction, indicating arterial thrombosis [3]. Another publication from the New England Journal, reported a young patient with headache, abdominal pain, anemia, and thrombocytopenia who had sinus vein thrombosis as a complication of PNH [4]. According to Beatrice Drexler, MD, of Basel University Hospital, thromboses are generally very relevant in patients with PNH because they affect mortality. They typically affect not only the deep leg veins, but can occur in quite atypical locations [1].
Flow cytometry as gold standard
The current gold standard for diagnostic confirmation is flow cytometry from peripheral blood, which detects the lack of expression of GPI-anchored molecules on erythrocytes and leukocytes. In this method, the cells to be examined flow one after the other through a thin measuring chamber, the so-called flow cell. Specific surface antigens of the cells are labeled with fluorescently labeled monoclonal antibodies to measure the fluorescence of each particle excited by the laser light. The minimum diagnostic criterion is a GPI-deficient population for at least two different GPI-anchored proteins on two different cell lines. Also relevant is the type and size of the PNH clone, which can also be defined in flow cytometry and provide information about the risk of thrombosis. Indications for flow cytometry include hemolysis, thrombosis, and bone marrow failure (Fig. 1) [5,6].
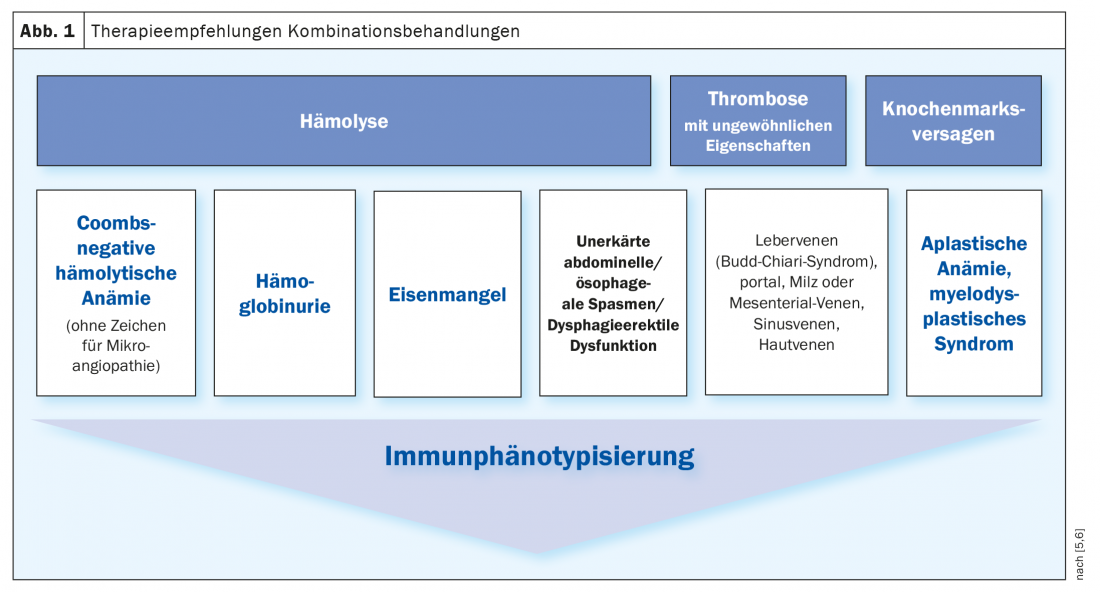
Therapy algorithm
In asymptomatic patients with unremarkable laboratory values, no treatment is usually required. However, these patients should be followed up regularly with blood counts, hemolysis parameters, and flow cytometry. In patients requiring treatment, the focus is on symptomatic therapy. A folic acid substitution is mandatory, as well as the administration of vitamin B12 and iron, depending on the laboratory values. Depending on the clone size and as needed, further supportive measures may be taken. In addition, inhibition of the terminal complement pathway is achieved by a monoclonal antibody such as eculizumab or ravalizumab. Infusion with these compliment inhibitors should occur every two or eight weeks. This can inhibit complement-mediated intravascular hemolysis, reduce transfusion requirements, improve patient quality of life, and also reduce the risk of thromboembolic events, which are the leading cause of death in PNH.
Take-Home Messages
- PNH is a heterogeneous disease with diverse manifestations.
- Often overlaps with other bone marrow disorders (aplastic anemia & myelodysplastic syndrome).
- The gold standard for diagnosis is flow cytometry from peripheral blood.
- Prognosis improved significantly with the administration of complement inhibitors.
- However, the therapy is very cost-intensive.
- Other disease-modifying therapies are in development.
Literature:
- Beatrice Drexler, MD, University Hospital Basel, “What if iron is deficient not because of iron?”, Iron Academy lecture, June 17, 2021.
- Parker, et al: Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005, doi: 10.1182/blood-2005-04-1717.
- Gerber, et al: Complement inhibition to treat myocardial infarction? BMJ Case Rep. 2011, doi: 10.1136/bcr.01.2011.3701.
- Sykes, et al: Case 40-2017. A 32-Year-Old Woman with Headache, Abdominal Pain, Anemia, and Thrombocytopenia. N Engl J Med. 2017, doi: 10.1056/NEJMcpc1710566.
- Röth, et al: Screening and Diagnostic Clinical Algorithm for Nocturnal Hemoglobinuria: Expert consensus. Eur J Haematol 2018, doi: 10.1111/ejh.13059.
- Borowitz, et al: Cytometry B Clin Cytome 78:211-230. Drexler B Pipette – Swiss Laboratory Medicine 2010, no 1/2 March 2021.
HAUSARZT PRAXIS 2021; 16(9): 20-22