Next to hematomatosis, cystic fibrosis (CF) is the most common genetic disease and the most common genetic disease with lung involvement. In adult patients, the symptoms are varied and often cannot be distinguished at first glance from other clinical pictures. A thorough medical history and the right tests help to track down the disease.
CF is no longer just a childhood disease – 51.2% of affected individuals on registries are in adulthood [1]. In Switzerland, about 1000 people are affected, recalled Dr. Macé Schuurmans, Department of Pneumology, University Hospital Zurich [2]. CF is inherited in an autosomal recessive manner, whereby there are about 2000 different variants of constellations regarding the hereditary changes. The affected gene encodes a multifunctional protein, CFTR (Cystic Fibrosis Transmembrance Regulator), which serves a chloride channel. There are severe mutations (class I-III) associated with pancreatic insufficiency. The most common mutation we have in Switzerland is shown here and concerns a deletion at site 508 (p.Phe508del) for phenylalanine in this gene. The pancreatic insufficiency results because on the one hand the patient has diabetes, but on the other hand he also has a digestive problem because the digestive enzymes are not functional.
CFTR modulator therapies have been available for approximately 5 years (ivacaftor, tezacaftor, lumacaftor, elexacaftor). The substances individually or in combination influence the function of the gene product both qualitatively and quantitatively. This innovation has led to a major advance in CF therapy, at least for those patients with the very common mutations.

Diagnosis
Newborn screening has been available since 2011 and is performed with an immunoreactive trypsinogen test (IRT) followed by DNA screening. Usually on the 4th day of life, a few drops of blood are taken from the child’s heel and placed on a filter paper card and analyzed in the screening laboratory. With this examination, 98% of all CF diseases are detected.
The advantages of testing as early as possible are obvious: “We know from various countries that have introduced screening earlier that the path to diagnosis is significantly shortened,” Dr. Schuurmans said. Thus, among other things, the parents could be advised regarding future pregnancies. The disease manifests itself less severely if treated properly and early, and lung function does not decline as rapidly.
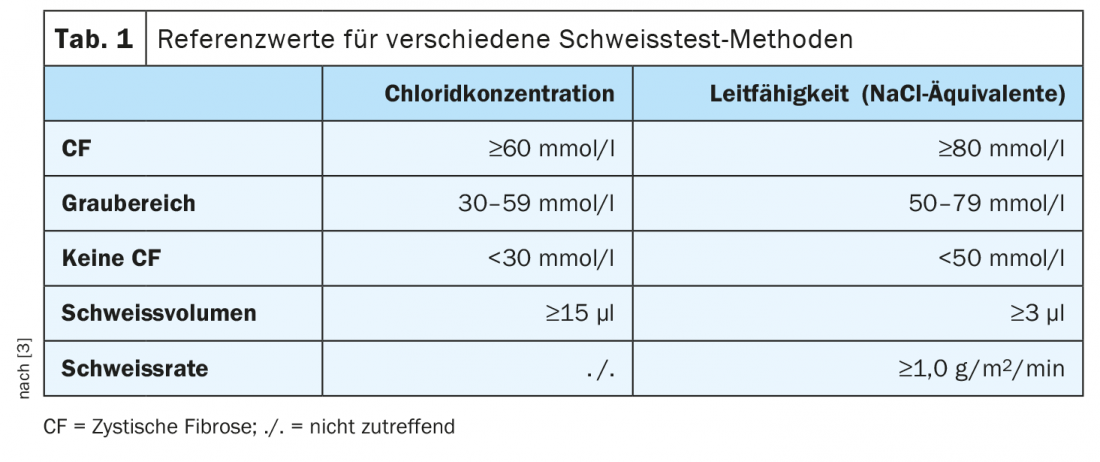
Prior to 2011, this screening was not available – all individuals born before then, as well as adults presenting with a late diagnosis, tend to have milder forms that are not necessarily as clear-cut in childhood. The most important first step when CF disease is suspected in this patient group is the sweat test, which is considered the gold standard in children born before 2011 as well as adults. The test determines both the chloride content and the conductivity. In a typical case, the chloride content is above 60 mmol/l, and a gray range exists between 30 and 59 mmol/l. If you are unsure about the diagnosis, you can use the conductivity as an aid. In contrast, if the value falls below 30 mmol/l, it is clear that CF is not present (Table 1) [3].
When to think about CF
In adults, CF should be considered if certain symptoms are present, and family history should also be obtained, Dr. Schuurmans advises. Symptoms include:
- recurrent respiratory infections, especially with certain “problem germs” such as Staph. aureus, Pseudomonas aeruginosa, Burkholderia cepacia complex or atypical mycobacteria (NTM).
- Atypical bronchial asthma, i.e., with a chronic productive cough that does not respond to standard therapy
- Bronchiectasis manifesting before the age of 40. “This can also happen later,” the expert said. “But before age 40, CF exclusion is mandatory.”
- Nasal polyposis, severe sinusitis
- Male infertility (can sometimes be the only sign in the early stages).
- Electrolyte disturbance (e.g. hyponatremia, dehydration)
- Acute pancreatitis
- Liver disease unexplained
If CF is suspected in adults, sweat testing or referral to an adult CF consultation is recommended first. The sweat test is classically performed at the children’s hospital. Genetics is only the second step, and it is mandatory that a counseling session then takes place by appropriately trained physicians.
If cystic fibrosis is not discovered until the age of 30, then survival is correspondingly longer. However, if you detect a patient early and their lung function is very good, then you have a correspondingly much longer time until either transplantation or the patient dies, Dr. Schuurmans summarized.
Congress: FomF WebUp Pneumology
Sources:
- Zolin A, Orenti A, Naehrlich L, et al: ECFSPR Annual Report 2018.
- FomF WebUp Pneumology, 7/12/2020.
- Young A: Switzerland Med Forum 2017; 17(24): 514-522.
HAUSARZT PRAXIS 2021; 16(2): 37 (published 2/19-21, ahead of print).
InFo PNEUMOLOGY & ALLERGOLOGY 2021; 3(1): 36.