With the aging of society, MDS diseases are also on the rise. However, developments in molecular diagnostics and new therapies will also change management for older patients. A multidisciplinary approach and exchange are important.
Myelodysplastic syndromes (MDS) are diagnosed as diseases of the elderly predominantly in patients >70 years. In Switzerland, with an incidence of 2-3/100,000 patient-years, slightly more than 300 new cases per year can be expected. There are currently estimated to be around 1600 patients with MDS living in our country [1]. Therapeutic options have remained essentially unchanged in recent years, with allogeneic stem cell transplantation as a curative treatment possible for only a few patients and various options to improve cytopenias in the remaining palliative situations [2–5]. In contrast, the rapid development of “next-generation” sequencing (NGS) has also led to relevant new findings in the clinical routine of hematology [6,7] that play a role for MDS patients with regard to diagnosis and prognosis assessment. Based on the updated WHO classification of 2016, the most important developments and concepts will be presented below.
Revision of the WHO classification 2016
Even in the age of molecular biology, the morphologic evaluation of peripheral blood smear and bone marrow aspirate remains the foundation of diagnosis. Basically, MDS forms with blast excess are distinguished from those without blast proliferation. The number of cell series affected by cytopenias and dysplasias, the detection of ring sideroblasts (RS) and typical cytogenetic alterations are decisive for further subdivision. A new feature of the 2016 WHO classification is the nomenclature (Table 1) [8,9]. The terms “refractory anemia” or “refractory cytopenia” have been abandoned and the term “myelodysplastic syndrome” is now used for all entities, supplemented by the core morphologic finding. This clears up some incongruities of previous terminology, such as the term “refractory anemia” for the forms of MDS with blast excess that are often accompanied by pancytopenia.

Conventional metaphase cytogenetics remains the second indispensable pillar of MDS diagnostics. In patients with cytopenias without blast proliferation and without (significant) dysplasias, the diagnosis can be made by detecting certain MDS-defining cytogenetic abnormalities (Table 2). In this case, the diagnosis of “unclassifiable MDS” is assigned. This category also includes cases with pancytopenia and single-line dysplasia or with constant blast detection of 1% in peripheral blood without blast proliferation in bone marrow [8,9].
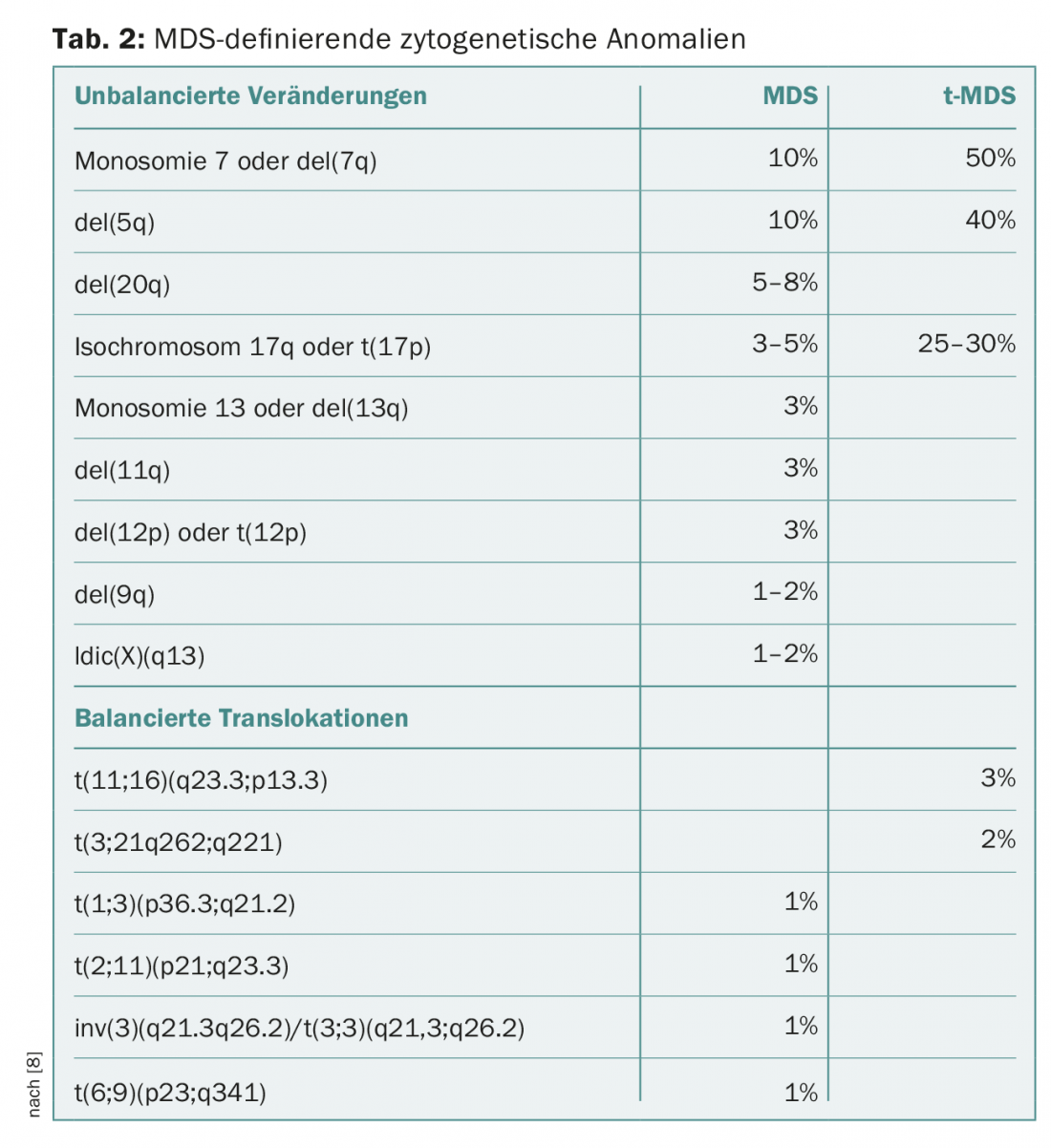
The category of MDS characterized by a deletion on the short arm of chromosome 5 with del(5q) has been expanded in its definition. Now, cases with a second cytogenetic abnormality can also be assigned to this category, except in the case of additional abnormalities in chromosome 7, which are associated with a significantly worse prognosis [8–10].
Conventional cytogenetics (analysis of at least 20 metaphases is required for a conclusive study) may be supplemented with other methods in selected situations, e.g., when MDS del(5q) is morphologically highly suspected but the karyotype is normal. Here, FISH panels focused on MDS-typical chromosomal abnormalities or genome-wide “comparative genomic hybridization” microarrays (array-CGH) [11] can be used. However, these two methods do not provide an a priori replacement for conventional cytogenetics. In particular, the prognostic significance of abnormalities detectable only in array CGH has not been clarified with certainty at present.
Diagnostic significance of “next-generation” sequencing in unclear cytopenias.
In recent years, numerous mutations have been identified in genes associated with the development of MDS and progression to AML. The spectrum of mutated “driver genes” is diverse and often includes components of the epigenetic modification of DNA and histones or the RNA-modifying splicing machinery (“spliceosome”). Components of cell cycle regulation, cohesin complexes, transcription factors, or components of intracellular signal transduction may also be affected (Table 3) [2,12–18].
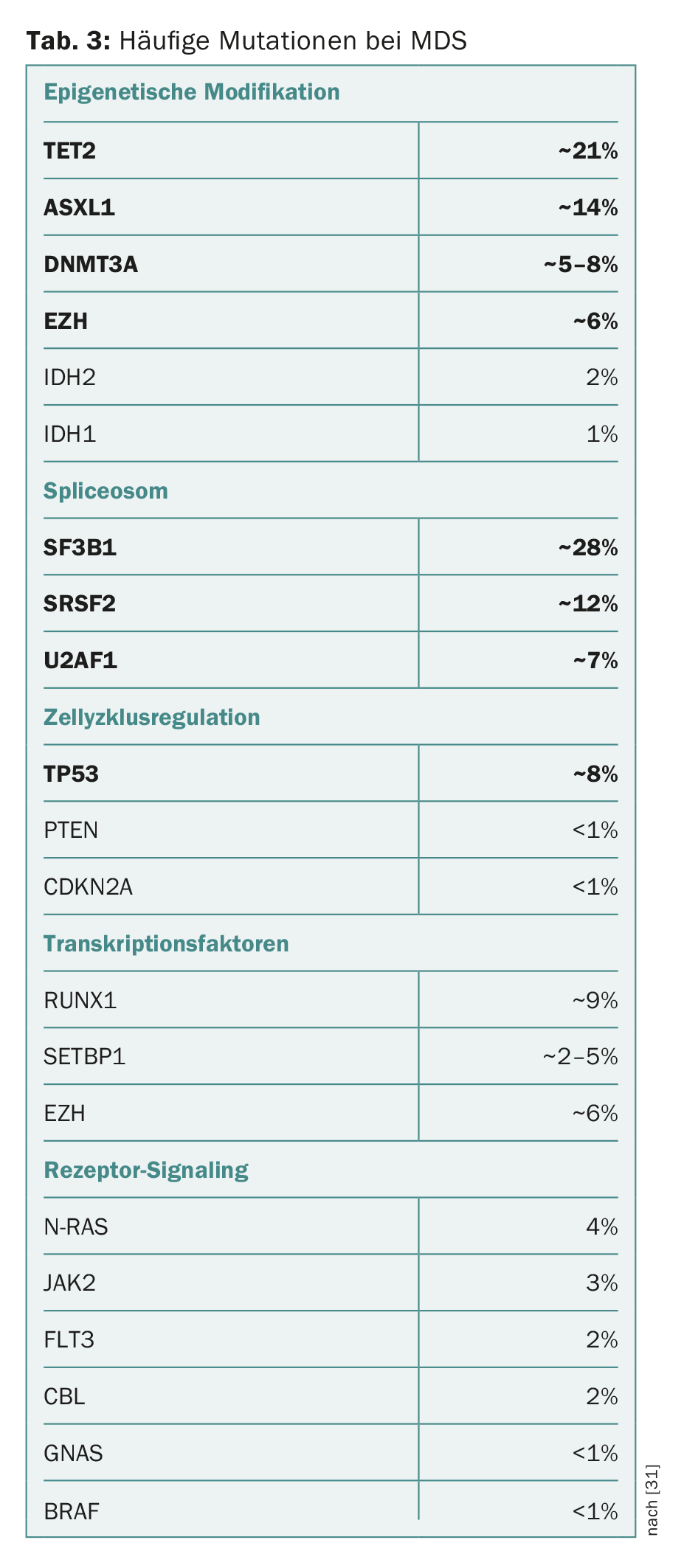
On the one hand, it should be emphasized that most of the driver gene mutations found in MDS also occur in other myeloid neoplasms, albeit in different frequencies or combinations. In addition, several studies have shown that mutations typical of myeloid neoplasms can also occur in hematologically healthy individuals. Incidence increases sharply with age, from 10% in 60-year-olds to 15-20% in >80-year-olds (but <1% in <40-year-olds). This phenomenon has been termed “Clonal Hematopoiesis of Indeterminate Potential” (CHIP) [19,20]. Similar to monoclonal gammopathy of uncertain significance (MGUS) and monoclonal B-cell lymphocytosis (MBL), it is a facultative precancerous condition that can progress to malignant hematologic disease at a rate of approximately 1% per year. In addition, patients with CHIP also have increased cardiovascular morbidity [21].
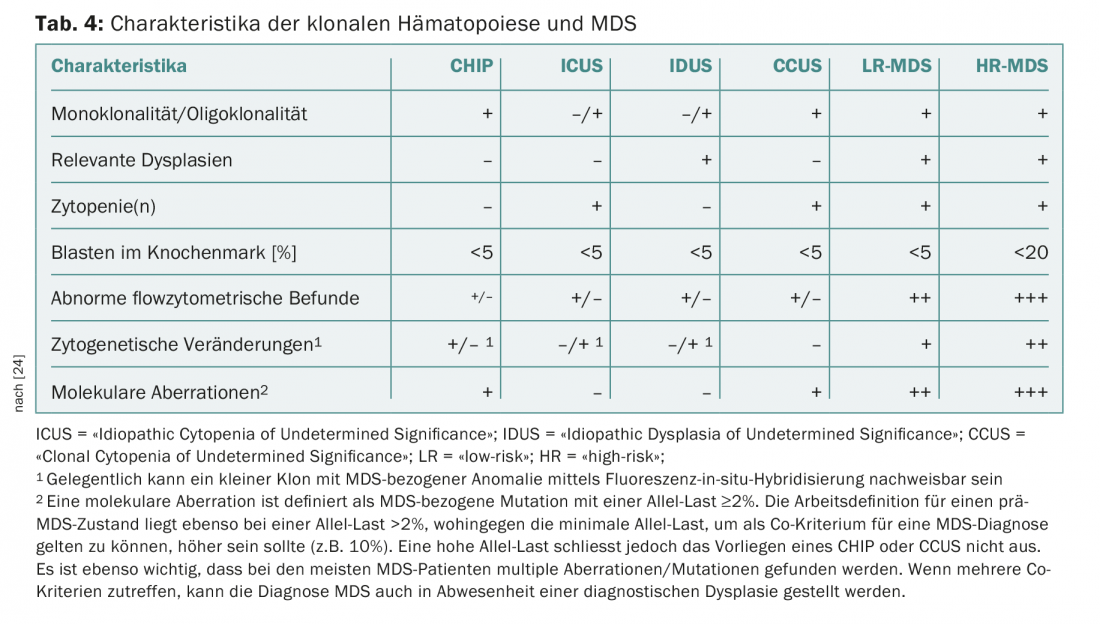
A recurrent problem in practice and clinic are patients with persistent cytopenias for which no definite causes can be found. In the absence of dysplasias or MDS-defining cytogenetic abnormalities, these have previously been grouped under the term “Idiopathic Cytopenia of Undetermined Significance” (ICUS) [22]. Detection of recurrent mutations may help to distinguish reactive from clonal cytopenias. The interpretation of a mutation detection depends on the size of the clone (measured as “variant allele frequency”, VAF) and the number of detected mutations. If a recurrent mutation with a VAF of more than 2% is found in an ICUS constellation, it is referred to as a “Clonal Cytopenia of Undetermined Significance” (CCUS). Among patients with CCUS, those with at least two mutations with a VAF >10% are at high risk of developing a hematologic neoplasm within the next five years [23]. The criteria recently presented by an international expert panel to differentiate CHIP, ICUS, CCUS from manifest MDS [24] are summarized in Table 4. This consensus also defines new MDS-related minor criteria that can be used to make a provisional MDS diagnosis in inconclusive situations (Table 5).

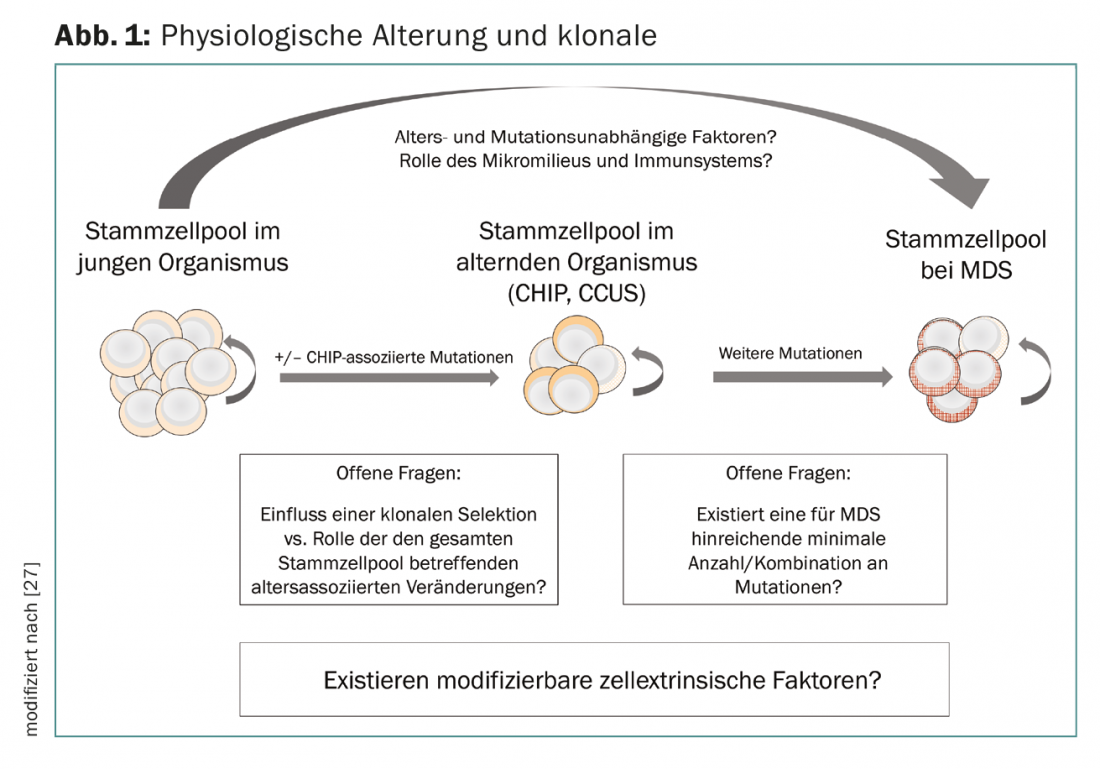
Sequential accumulation of genetic alterations in HSCs has long been postulated as a pathogenetic correlate of clonal evolution, and the concept of clonal hematopoiesis provides evidence for an overlap between alterations in hematopoiesis with age and the pathogenesis of myeloid neoplasms. However, it remains unclear what factors are responsible for the transition of CHIP and CCUS to manifest neoplasia. In this context, clonal evolution seems to be conditioned not only by cell-intrinsic (mutations in hematopoietic stem cells) but also by cell-extrinsic mechanisms. In this regard, the bone marrow microenvironment and components of the innate and acquired immune system play a role. The “immunologic stress” probably also explains the association with sometimes unclassifiable inflammatory and immunologic concomitant diseases or phenomena that may occur in patients with MDS [25–28]. The loss of immunological tumor control and also the facilitating changes in the bone marrow niche are currently the focus of basic research. This will potentially lead to new therapeutic approaches in the future that could be used early in the development of myeloid neoplasms (Fig. 1).
Due to the overlap in the mutation spectrum between CHIP, CCUS and the MDS, mutation analyses were deliberately not included in the current WHO classification. An exception is mutations in the driver gene of the spliceosome component SF3B1, which are strongly associated with a ringsideroblastic phenotype [29]. According to WHO 2016, in the case of an SF3B1 mutation, detection of 5% RS is sufficient for classification in the group of MDS with RS, instead of the 15% otherwise required. SF3B1-mutated patients have a very good prognosis with low probability of progression to AML. Because patients with multilineage dysplasia and RS also benefit from the prognostic impact of an SF3B1 mutation, the entity MDS with multilineage dysplasia and RS was reincluded in the 2016 classification.
Further prognostic and predictive significance of driver gene mutations.
From a prognostic point of view, some further clinical scenarios can already be named in which mutation analyses can provide relevant information. Approximately 15% of patients with MDS del(5q) have a TP53 mutation. These also respond hematologically to lenalidomide, but are less likely to achieve cytogenetic remission and are at higher risk for AML transition. Therefore, when a TP53 mutation is detected in MDS del(5q), treatment alternatives can be considered [3,30].
Another scenario for mutation search concerns the heterogeneous group of patients in the “intermediate” group according to IPSS-R. These can be treated according to either the recommendations applicable to “low-” or “high-risk” patients, depending on the presence or absence of other risk features [2–5,30]. So far, only conventional additional risk markers (e.g. LDH elevation, bone marrow fibrosis >grade 2 according to WHO) are available for this purpose besides the isolated examination of cytogenetics (high-risk constellation?). Several recurrent mutations are associated with significantly increased risk and warrant prognostic upstaging to the “high-risk” category and, in selected patients, allogeneic stem cell transplantation [15]. Therefore, searching for TP53, ASXL1, RUNX1, EZH2, and ETV6 mutations in patients who qualify for intensive therapy is explicitly recommended in the current guidelines [4,15]. An international project sponsored by the MDS Foundation is currently striving to develop a “molecular IPSS-R” supplemented by mutational status.
Clinically useful data on the predictive value of mutation profile in MDS are currently limited. Relevance to stratification with respect to targeted therapies is emerging for the future. Luspatercept (ACE-356) is a TGF-β superfamily inhibitor [10], which has shown a high erythroid response rate in EPO-refractory MDS patients with RS and/or mutations in SF3B1. In addition, spliceosome inhibitors (H3B-8800) are currently being investigated in AML and MDS that preferentially eliminate clones with mutations in spliceosome driver genes due to haplo-insufficiency (cyclops effect). In addition, midostaurin (FLT3 mutations), enasidenib (IDH2 mutations) and ivosidenib (IDH1 mutations), which are already approved for AML, are currently in clinical development for high-risk MDS with a corresponding mutation profile, either as a single agent or in combination with hypomethylating therapy (HMT) or standard chemotherapy.
Take-Home Messages
- Due to the aging of our society, a significant increase in MDS disease is expected.
- Developments in molecular diagnostics as well as new therapeutic options will change treatment strategies also for elderly patients.
- Multidisciplinary management is an important requirement and poses new challenges for health systems. This concerns not only adequate care in routine clinical practice, but also the conduct of clinical trials, which require a high degree of cooperation and coordination in rare diseases in the age of personalized medicine.
- In response to these challenges, the Swiss MDS Study Group launched the Swiss MDS Registry/Biobank in 2015 to enable clinical and scientific exchange within an international network.
Literature:
- Bonadies N, et al: Trends of classification, incidence, mortality, and survival of MDS patients in Switzerland between 2001 and 2012. Cancer epidemiology 2017; 46: 85-92.
- Malcovati L, et al: Diagnosis and treatment of primary myelodysplastic syndromes in adults: Recommendations from the European LeukemiaNet. Blood 2013; 122(17): 2943-2964.
- Fenaux P, et al: Myelodysplastic syndromes: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology: official journal of the European Society for Medical Oncology 2014; 25(Suppl 3): iii57-69.
- Hofmann WK, Platzbecker U, Götze K: Onkopedia guideline myelodysplastic syndromes. As of March 2016.
- Greenberg PL, et al: Myelodysplastic Syndromes, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network: JNCCN 2017; 15(1): 60-87.
- Johnsen JM, Nickerson DA, Reiner AP: Massively parallel sequencing: the new frontier of hematologic genomics. Blood 2013; 122(19): 3268-3275.
- Kuo FC, et al: The relative utilities of genome-wide, gene panel, and individual gene sequencing in clinical practice. Blood 2017; 130(4): 433-439.
- Swerdlow SH, et al. (Eds.): WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th edition. Lyon: International Agency for Research on Cancer 2017.
- Arber DA, et al: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127(20): 2391-2405.
- Mies A, Platzbecker U: Increasing the effectiveness of hematopoiesis in myelodysplastic syndromes: erythropoiesis-stimulating agents and transforming growth factor-β superfamily inhibitors. Seminars in hematology 2017; 54(3): 141-146.
- Ouahchi I, et al: Microarray-based comparative genomic hybridisation reveals additional recurrent aberrations in adult patients evaluated for myelodysplastic syndrome with normal karyotype. British journal of haematology 2018. DOI: 10.1111/bjh.15068 [Epub ahead of print].
- Kon A, et al.: Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nature genetics 2013; 45(10): 1232-1237.
- Yoshida K, et al: Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478(7367): 64-69.
- Bejar R, Levine R, Ebert BL: Unraveling the molecular pathophysiology of myelodysplastic syndromes. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2011; 29(5): 504-515.
- Bejar R, et al: Clinical effect of point mutations in myelodysplastic syndromes. The New England journal of medicine 2011; 364(26): 2496-2506.
- Abdel-Wahab O, Figueroa ME: Interpreting new molecular genetics in myelodysplastic syndromes. Hematology American Society of Hematology Education Program 2012; 2012: 56-64.
- Leeke B, et al: Cohesin mutations in myeloid malignancies: Underlying mechanisms. Experimental hematology & oncology 2014; 3: 13.
- Tothova Z, Steensma DP, Ebert BL: New strategies in myelodysplastic syndromes: Application of molecular diagnostics to clinical practice. Clinical cancer research: an official journal of the American Association for Cancer Research 2013; 19(7): 1637-1643.
- Jan M, Ebert BL, Jaiswal S: Clonal hematopoiesis. Seminars in hematology 2017; 54(1): 43-50.
- Heuser M, Thol F, Ganser A: Clonal Hematopoiesis of Indeterminate Potential. Deutsches Arzteblatt international 2016; 113(18): 317-322.
- Fuster JJ, Walsh K: Somatic Mutations and Clonal Hematopoiesis: Unexpected Potential New Drivers of Age-Related Cardiovascular Disease. Circulation research 2018; 122(3): 523-532.
- Valent P, et al: Idiopathic cytopenia of undetermined significance (ICUS) and idiopathic dysplasia of uncertain significance (IDUS), and their distinction from low risk MDS. Leukemia research 2012; 36(1): 1-5.
- Malcovati L, et al: Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017; 129(25): 3371-3378.
- Valent P, et al: Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget 2017; 8(43): 73483-73500.
- Gañán-Gómez I, et al: Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia 2015; 29(7): 1458-1469.
- Glenthøj A, et al: Immune Mechanisms in Myelodysplastic Syndrome. International journal of molecular sciences 2016; 17(6): 944.
- Chung SS, Park CY: Aging, hematopoiesis, and the myelodysplastic syndromes. Blood advances 2017; 1(26): 2572-2578.
- Cooper JN, Young NS: Clonality in context: hematopoietic clones in their marrow environment. Blood 2017; 130(22): 2363-2372.
- Malcovati L, et al: SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015; 126(2): 233-241.
- Nordic MDS Study Group: Guidelines to Patient Management of Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia. 8th update. www.nmds.org/index.php/guidelines (cited Mar. 14, 2018).
- Montalban-Bravo G, Garcia-Manero G: Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am J Hematol 2018 Jan; 93(1): 129-147.
Further reading:
- List A, Ebert BL, Fenaux P: A decade of progress in myelodysplastic syndrome with chromosome 5q deletion. Leukemia 2018. DOI: 10.1038/s41375-018-0029-9 [Epub ahead of print].
- Platzbecker U, et al: Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): A multicentre, open-label phase 2 dose-finding study with long-term extension study. The Lancet Oncology 2017; 18(10): 1338-1347.
InFo ONCOLOGY & HEMATOLOGY 2018; 6(2): 22-26.