The systemic autoimmune disease APS (antiphospholipid syndrome) is mainly characterized by thrombotic manifestations. As acquired thrombophilia, variable drug thromboprophylaxis is indicated.
Antiphospholipid syndrome (APS) is a systemic autoimmune disorder characterized by the occurrence of venous and/or arterial thrombosis and/or pregnancy complications [1]. The pathogenesis of APS is based on the presence of antibodies directed against a variety of phospholipid/protein complexes [2]. The discovery and knowledge of these components spans several decades (Table 1).

The pathomechanism of how antiphospholipid antibodies (aPL) lead to coagulation activation is multifactorial (Table 2). There are different approaches from human and animal studies demonstrating the involvement of aPL [2]. APS can occur in isolation (primary APS) or in the setting of other autoimmune diseases (e.g., SLE, malignancies). A rare but very severe course of APS is catastrophic antiphospholipid syndrome with simultaneous involvement of three or more organ systems [3].

Antiphospholipid antibodies (aPL)
Antiphospholipid antibodies are a heterogeneous group of acquired antibodies directed against phospholipid-protein complexes and showing a high affinity for negatively charged surfaces. Major target antigens (proteins) include β2-glycoprotein-I (β2-GPI) and prothrombin. Other antigens such as activated protein C, protein S, annexin V, oxidized LDL, and factor XII have also been described (Fig. 1) [2,4]. Interactions of aPL on negatively charged phospholipid surfaces cause various responses such as inhibition of hemostatic regulation (by inhibition of protein C or antithrombin, or displacement of annexin V from throphoblasts in placenta), activation of platelets (by stimulation of thromboxane A2), upregulation of adhesion molecules and tissue factor on endothelial cells, and complement activation. (Tab.2).
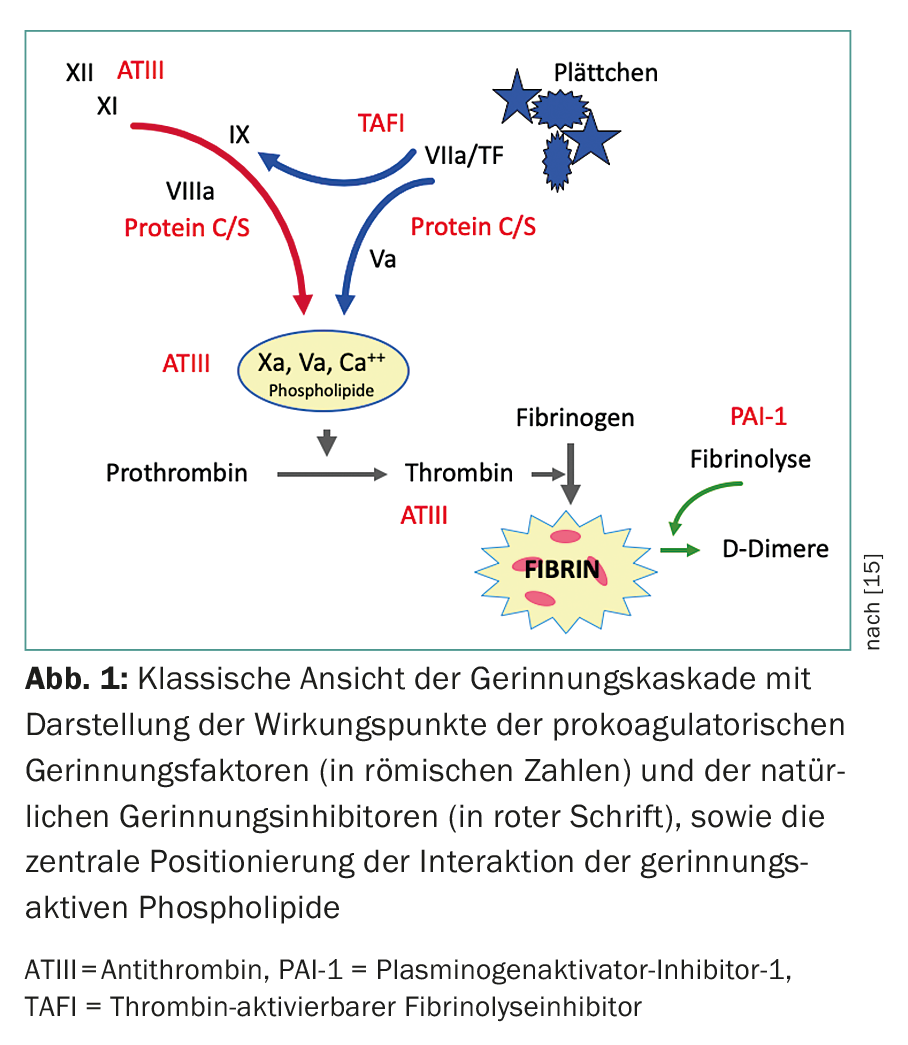
aPL can lead to a prolongation of the clotting time in phospholipid-dependent coagulation tests (e.g. Quick/INR or APTT), because they interfere with or block the phospholipids of the prothrombinase complex, which are necessary for the course of the coagulation cascade. This in vitro phenomenon led to the misleading name “lupus anticoagulant” in 1952, when this “anticoagulant” effect was observed in patients with lupus erythematosus in laboratory tests.
Classification criteria
The GSP classification criteria were first formulated during a workshop in Sapporo (Japan) in 1998 and revised in 2005 during the “11th International Congress on Antiphospholipid Antibodies” in Sydney [5,6]. Accordingly, a clinical criterion (venous and/or arterial thrombosis in the microcirculation or macrocirculation and obstetric complications) plus a laboratory criterion (twice positive at 12 weeks for lupus anticoagulant, anticardiolipin antibody, or anti-β2GPI antibody) are required to define definite APS (Table 3).
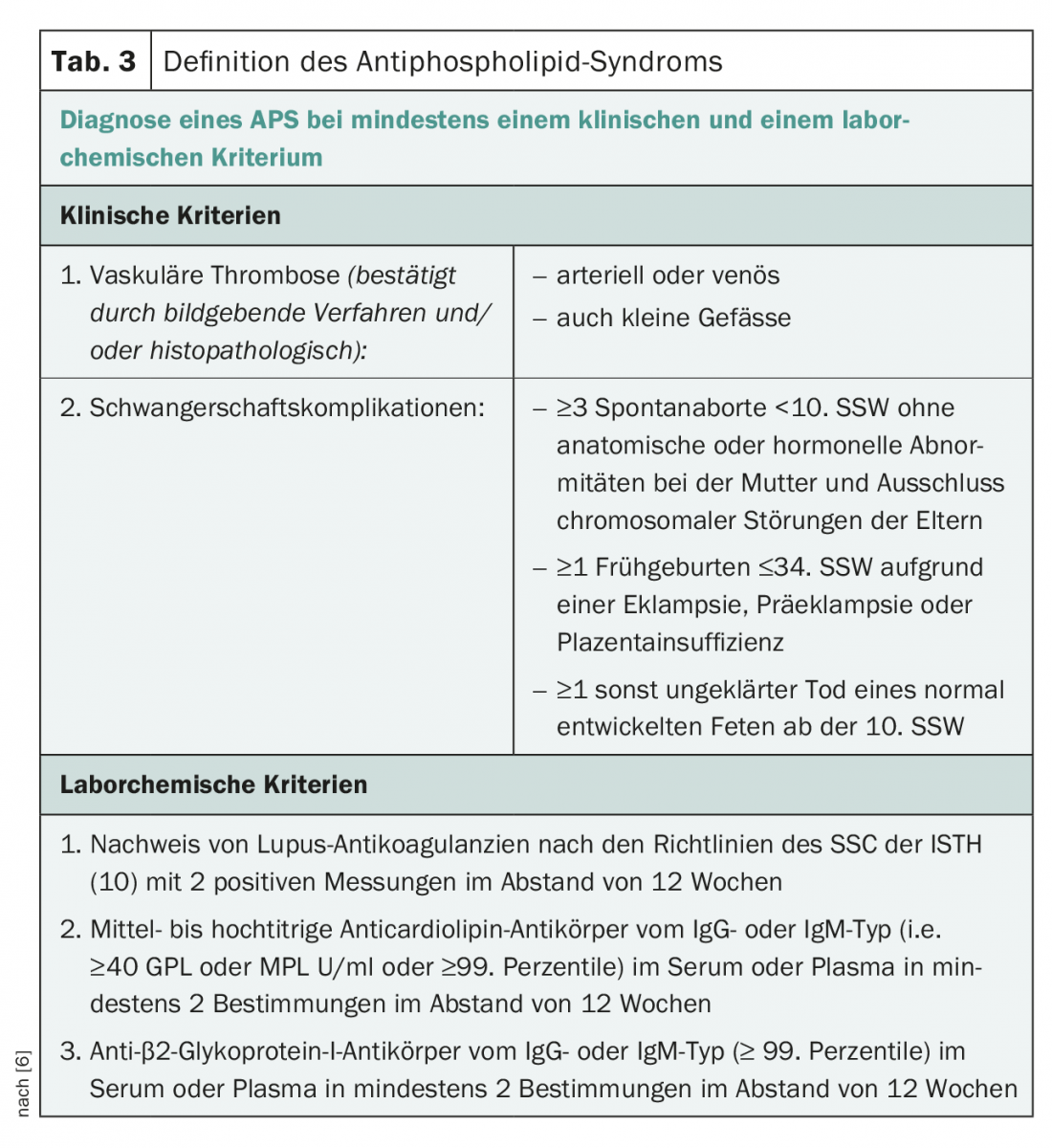
Diagnostics of the GSP
For the detection of aPL (LA, aCL-IgG /-IgM, β2GPI-IgG/-IgM), several test methods are combined according to international recommendations [7,8]. Basically, lupus anticoagulants are functionally identified by coagulation tests. aCL and β2GPI antibodies, on the other hand, are detected immunologically, by ELISA or chemiluminescence assay. Determination of aPL twice at intervals of at least twelve weeks is required to avoid detection of any transient antibodies that may occur, for example, in the context of infections. Figure 2 illustrates the diagnostic algorithm, which is intended to illustrate the complexity of the diagnostic steps in cases of suspected APS.
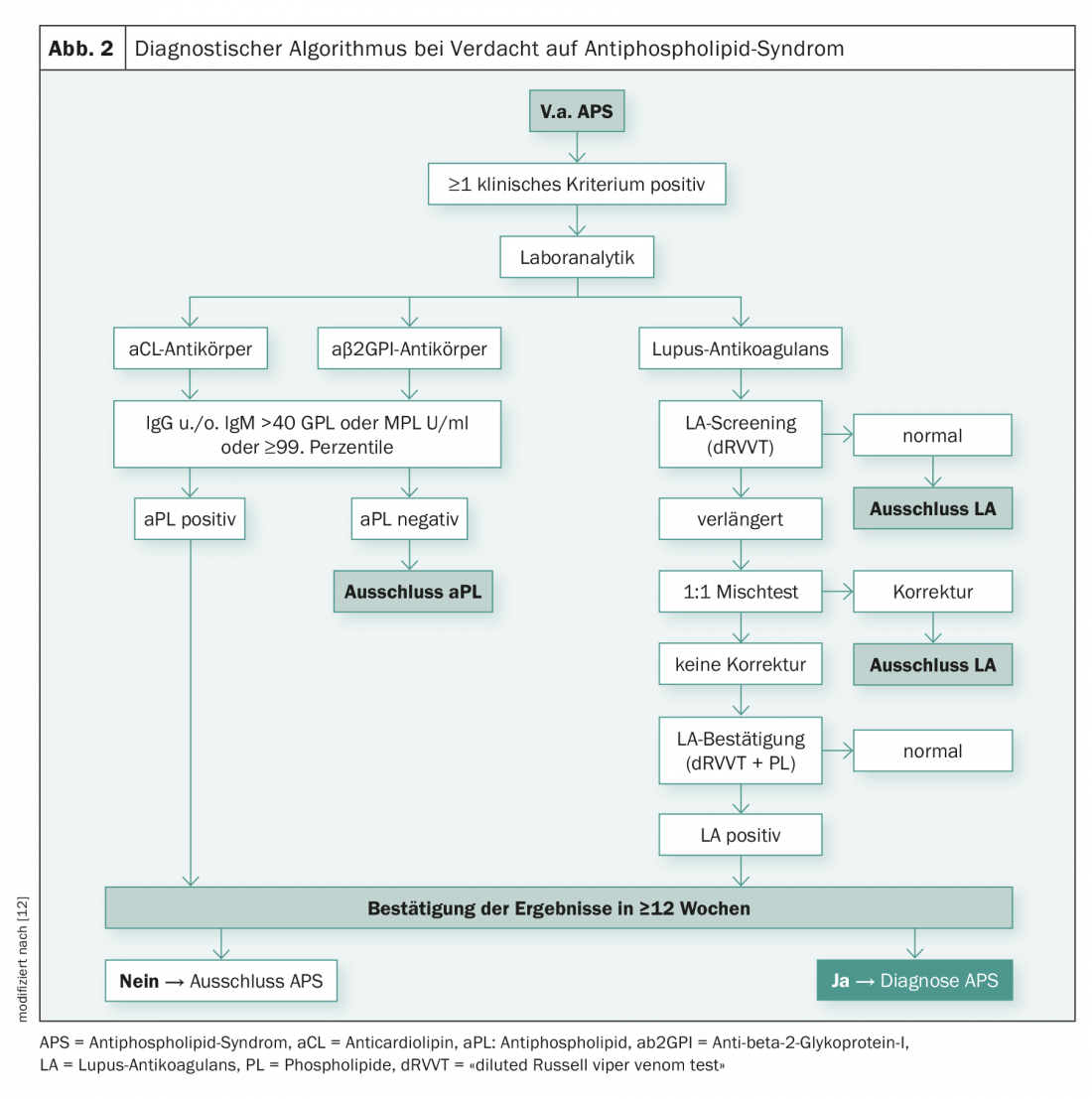
Coagulation testing – Lupus anticoagulant: The International Society of Thrombosis and Hemostasis (ISTH) defines a three-tiered strategy in each of two different LA testing procedures [7]:
- Screening test: prolongation of a phospholipid-dependent clotting time (>99th percentile);
- Mixed test: Confirmation of an immediate-acting inhibitor and exclusion of coagulation factor deficiency;
- Confirmatory test: Confirmation that the inhibitor is phospholipid-dependent. The third step is necessary to ensure that the inhibitor is not directed against a specific clotting factor.
Immunological tests – aCL/aβ2GPI-ELISA: In addition to the coagulation tests, aPL antibodies (aCL-IgG and -IgM as well as aβ2GPI-IgG and -IgM) are determined immunologically by ELISA. In view of the variable antigen properties as well as the lack of reference materials, standardization of test systems is very difficult. IgG and IgM antibody titers are expressed in internationally standardized GPL and MPL units. Positive results are defined as titers ≥40 GPL/MPL units (U/ml) or ≥99th percentile of the respective method [8].
Although anti-β2GPI assays have greater specificity with respect to thrombogenicity than aCL, they are also not yet sufficiently standardized. Test methods that use the recombinant domain 1 of β2GPI, i.e. the epitope for aPL binding, as antigen are considered promising [9,10]. Recent work has shown that antibodies directed against this epitope are more strongly associated with clinical symptoms. However, further efforts are needed to achieve improved standardization of the different test methods as well as better characterization of clinically relevant aPL.
Clinic
APS is a multi-organ disease with multiple manifestations, which is classified as an acquired thrombophilia. The clinical picture is characterized by recurrent arterial and/or venous thromboses of small or large vessels with typical or atypical localization, as well as vascular pregnancy complications. In addition, other clinical manifestations have been described, such as neurological dysfunction (epilepsy, dementia), dermatological symptoms (livedo reticularis/racemosa, acral necrosis), valvular heart disease (non-bacterial thrombotic endocarditis) and myocardial infarction, Renal diseases (renal artery/renal vein thrombosis, thrombotic microangiopathy), ocular diseases (amaurosis fugax, occlusions of retinal and/or choroidal vessels), and thrombocytopenias. It is not always clear in these clinical pictures whether the aPL represent an epiphenoma or are pathogenetically involved.
Indication: aPL screening should be initiated if there is a high probability of APS based on a clinical criterion (Tab. 3) . Untargeted aPL screening in asymptomatic individuals is not recommended given the low specificity of the test systems.
The diagnosis of APS is based on repeated detection of LA, aCL, or aβ2GPI at 12-week intervals, as mentioned above. Based on the aPL profile (isotype and number of positive tests), both a risk assessment and an estimation of the reliability of the results can also be made [11]. Thus, it was shown that 98% of triple positive, 84% of double positive and 40% of single positive patients still tested positive after twelve weeks. These data underscore that the triple positive profile is a robust laboratory result. In addition, a triple positive profile corresponds to a classification for high-risk APS, whereas a double or single positive profile corresponds to moderate or low risk for APS, respectively.
Therapy
The treatment strategy when aPL is detected depends primarily on whether the patients are asymptomatic with only positive laboratory findings or symptomatic patients with manifest complications. In asymptomatic patients, the same recommendation for thromboprophylaxis applies as for other thrombophilic patients. In the case of manifest APS with corresponding symptoms, however, a distinction must be made as to whether the primary or secondary form is present. In primary APS without evidence of another underlying disease, therapy is directed solely at preventing further complications of thrombosis. In secondary APS, the first step is to treat the underlying disease or minimize the typical risk factors for cardiovascular complications, in parallel with treatment of the thrombotic syndrome.
Anticoagulation (ASA, VKA, NOAK): In APS patients with venous thrombosis, initial therapy with unfractionated (UFH) or low-molecular-weight (NMH) heparin followed by long-term therapy with vitamin K antagonists (VKA) is recommended. Because of the high risk of recurrent thrombosis (depending, among other things, on whether the patient belongs to the high-risk group with triple positive aPL findings) after discontinuation of oral anticoagulation, long-term oral anticoagulation with target INR 2-3 is the therapy of choice in most cases, at least as long as laboratory findings remain positive [12]. This significantly reduces thromboembolic recurrences. Nevertheless, it turns out to be insufficient in some cases. In such cases, long-term therapy with NMH or raising the target INR to 3-4 may be considered.
Patients with arterial thrombosis or triple-positive APS patients who are at high risk for thromboembolic events benefit more from systemic anticoagulation than from antiplatelet agents. Lifelong anticoagulation is indicated in such patients.
The use of the new direct oral anticoagulants (NOAKs) is controversial [12]. NOAKs do not require laboratory controls and have been found to be at least as effective and safe as VKA or NMH in the treatment or prevention of venous and arterial thromboembolism. However, prospective and retrospective studies have demonstrated that NOAKs are less effective than warfarin in high-risk APS patients [13,14]. The current evidence base for the use and efficacy of NOAKs in APS patients is still insufficient, and additional prospective studies are needed to assess their value in APS.
Anticoagulation in women with recurrent pregnancy complications due to APS is aimed at fetal preservation or increased live birth rate. For the treatment of pregnant women, a combination of NMH in sub-therapeutic dosage (1×100 E/kgKG/day) and aspirin (1×100 mg/day) is recommended. Anticoagulation should be started as soon as possible after pregnancy and continued until six weeks postpartum.
Immunosuppression: Treatment with immunosuppressive agents in addition to anticoagulation is indicated especially in the presence of secondary APS, thrombocytopenia, or catastrophic antiphospholipid syndrome (CAPS). The use of immunosuppressants in APS for prophylaxis of further thromboembolic events is also discussed, because immunosuppression has a favorable influence on high aPL titers and thus could possibly result in a reduced incidence of vascular occlusion. However, specific recommendations regarding the type and duration of immunosuppression are not available. These are derived from experience with treatment of autoimmune thrombocytopenia or coagulation factor inhibitors (e.g., single or combination therapy with prednisone/cyclophosphamide/rituximab).
Take-Home Messages
- Antiphospholipid syndrome (APS) is a systemic autoimmune disease with primary pathogenetic event: formation of autoantibodies against coagulase-active phospholipid/protein complexes.
- Despite the paradoxical phenomenon of prolongation of global coagulation tests in vitro, the clinical picture of the patient with APS is characterized by thrombotic manifestations rather than bleeding.
- The diagnosis of APS requires laboratory findings (lupus anticoagulant and/or medium-high positive antiphospholipid antibodies) combined with clinical thrombosis in the micro- or macrocirculation.
- APS is considered an acquired thrombophilia and needs variable drug thromboprophylaxis as therapy, adjusted for intensity and duration, depending on organ involvement and extent of thrombotic manifestations. Heparins or vitamin K antagonists remain first-line anticoagulants.
Literature:
- Cervera R, Espinosa, Khamashta MA: Antiphospholipid syndrome in systemic autoimmune diseases, 2nd edition. Elsevier, Amsterdam, 2016.
- Giannakopoulos B, Krilis SA: The pathogenesis of the antiphospholipid syndrome. N Engl J Med 2013; 368: 1033-1044.
- Asherson RA, et al: Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus 2003; 12: 530-534.
- Arnout J, Vermylen J: Current status and implications of autoimmune antiphospholipid antibodies in relation to thrombotic disease; J Thromb Heamost 2003; 1: 931-942.
- Wilson WA, et al: International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum 1999; 42: 1309-1311.
- Miyakis S, et al: International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4: 295-306.
- Pengo V, et al: Update of the guidelines for lupus anticoagulant detection. Subcommittee on lupus anticoagulant/antiphospholipid antibody of the scientific and standardisation committee of the international society on thrombosis and haemostasis, J Thromb Haemost 2009; 7: 1737-1740.
- Devreese KMJ, et al:. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibodies, Laboratory criteria for antiphospholipid syndrome: communication from the SSC of the ISTH, J Thromb Haemost 2018; 16: 809-813.
- De Craemer AS, Musial J, Devreese KM: Role of anti-domain 1-beta2 glycoprotein I antibodies in the diagnosis and risk stratification of antiphospholipid syndrome. J Thromb Haemost 2016; 14: 1779-1787.
- Pengo V, et al: Antiphospholipid syndrome: antibodies to domain 1 of beta2-glycoprotein 1 correctly classify patients at risk. J Thromb Haemost 2015; 13:782-7.
- Pengo V, et al: Confirmation of initial antiphospholipid antibody positivity depends on the antiphospholipid antibody profile. J Thromb Haemost 2013; 11: 1-5.
- Garcia D, Erkan D: Diagnosis and Management of the Antiphospholipid Syndrome. N Engl J Med 2018; 378: 2010-2021.
- Martinelli I, et al: Recurrent thrombosis in patients with antiphospholipid antibodies treated with vitamin K antagonists or rivaroxaban. Haematologica 2018;103(7): 315-317.
- Pengo V, et al: Rivaroxaban vs warfarin in high-risk patients with antiphospholipid syndrome. Blood 2018; 132(13): 1365-1371.
- Tsakiris D, Bachofner A: Disseminated intravascular coagulation in the tumor patient. Info@Oncology 2017; 7:19-21.
InFo ONCOLOGY & HEMATOLOGY 2019; 7(1): 22-25.