The term indolent lymphomas encompasses a growing number of low-malignant non-Hodgkin lymphomas and leukemias predominantly of the B-cell series. They are differentiated and classified from each other based on their marker constellation and genetic profile. Treatment is based on the extent or dynamics of the disease and the patient’s symptoms. There is currently a shift in therapy from classical chemotherapeutics to signal inhibitors and immunotherapeutics. The primary goal in treatment is often to control the disease for as long as possible with acceptable tolerability of therapy.
The term indolent non-Hodgkin’s lymphoma (iNHL) encompasses a biologically heterogeneous group of mature and small cell lymphomas of the B-cell series with a tendency to leukemic manifestation. In the past, the term “indolent” described a group of slow-growing, “low-malignant” lymphomas whose subtypes were often difficult to separate from one another and which did not always need to be differentiated from one another due to a lack of therapeutic options.

Meanwhile, advanced immunophenotypic and molecular data allow a better subdivision into lymphoma entities, which should be done according to the current WHO lymphoma classification 2008 (Table 1) [1]. There are iNHL that transform into highly malignant blastic or aggressive lymphoma during the course of the disease, most notably follicular lymphoma, followed by chronic lymphocytic leukemia (CLL). In addition, there are entities such as mantle cell lymphoma (MCL), the majority of which (approximately 90% of all cases) exhibit aggressive growth behavior and are treated with intensive immunochemotherapy (with high-dose therapy and autologous stem cell replacement if necessary). But then there is also an indolent subgroup that usually manifests in older patients with bone marrow and spleen involvement. Not to forget the typical NHL that may occur in a specific clinical context such as Helicobacter pylori-associated MALT or HCV-associated marginal zone lymphoma.
Diagnosis and subtypes
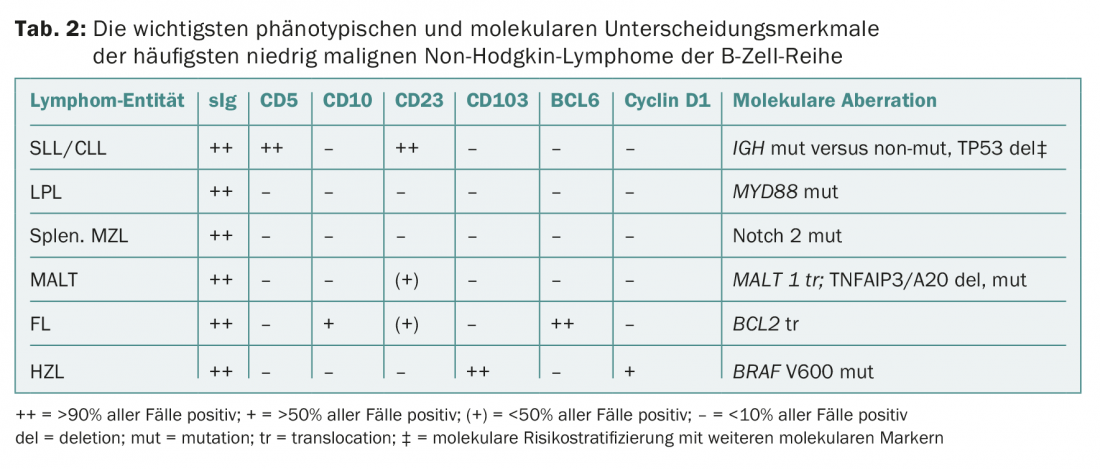
Diagnosis is made by lymph node and/or bone marrow biopsy, (immune) phenotyping of blood and/or bone marrow, and genetic-molecular analyses (Table 2) . These tests are standardized and apply to all lymphoma pathologies. According to the WHO classification, the following entities belong to iNHL:
- Chronic lymphocytic leukemia (CLL) resp. its nodal form (small cell lymphocytic lymphoma, SLL)
- Lymphoplasmacytic lymphoma (LPL or Waldenström’s disease)
- Marginal zone lymphoma (MZL) (Fig. 1) with its three subtypes: nodal, extranodal (e.g., as MALT), and splenic
- Follicular lymphoma (FL) (Fig. 2)
- Hairy Cell Leukemia (HZL).
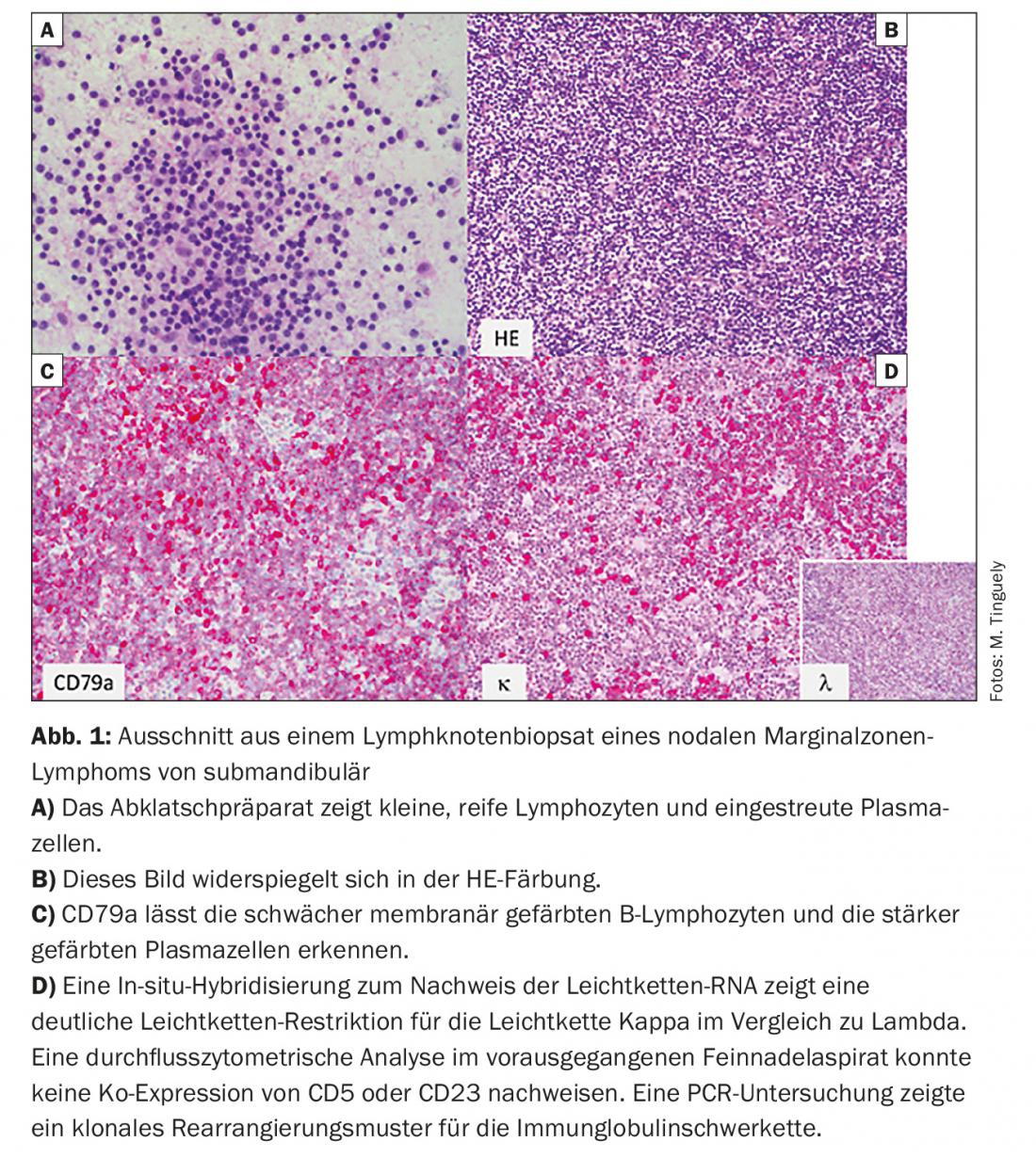
CLL and hairy cell leukemia are not considered by all research groups to be part of iNHL because, as their names suggest, they often present primarily with blood picture changes and in some cases (this applies in particular to hairy cell leukemia) require different therapeutic measures. Mantle cell lymphoma (MCL) should no longer be included in iNHL for the reasons stated, although there is an indolent subgroup in elderly patients.
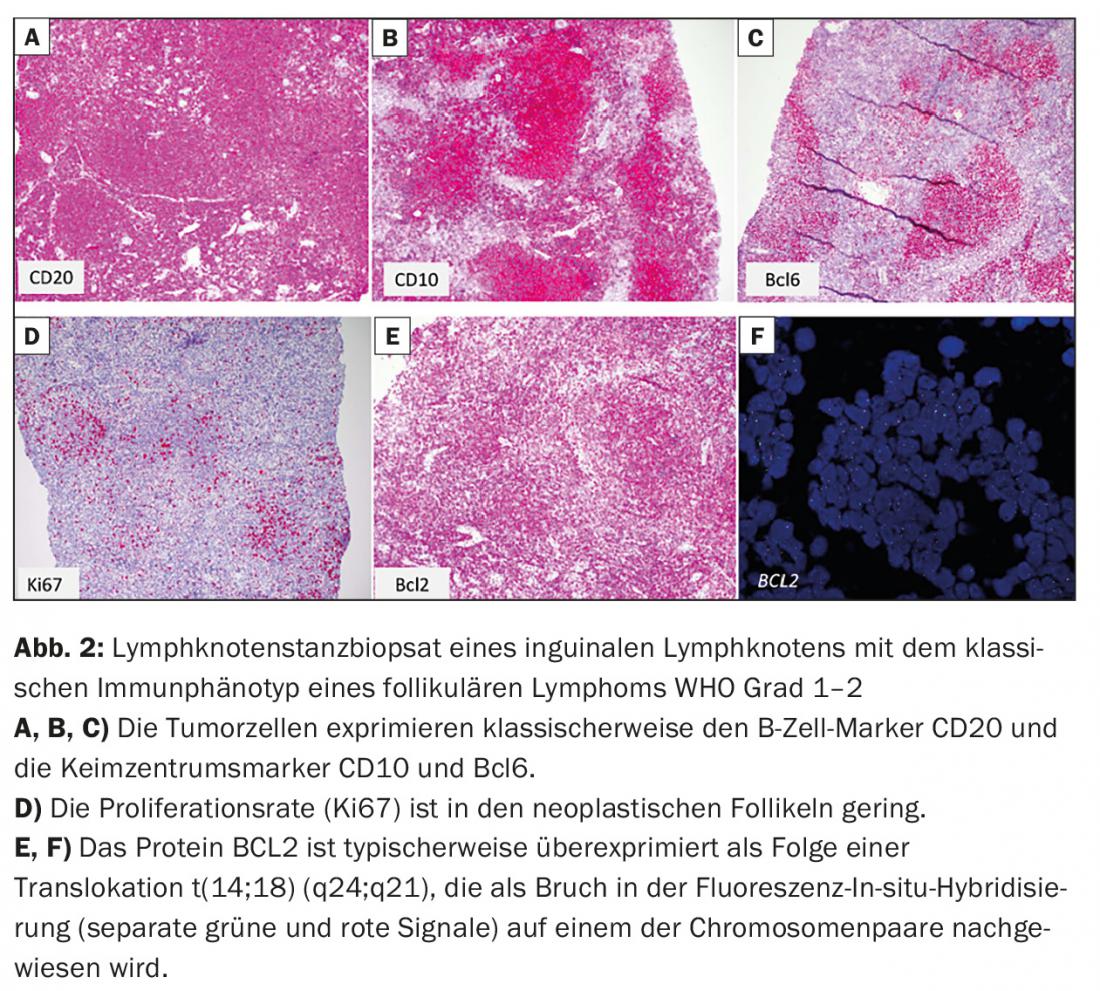
Clinical presentation and therapeutic principles
Indolent lymphoma is usually a disease of advanced age. The vast majority of patients (66%) are already in advanced stages III and IV (staging according to Ann Arbor or, more recently, Lugano classification) at the time of diagnosis and are thus no longer amenable to curative therapy [2]. The diagnosis is not always associated with the need to initiate therapy immediately. Although recommendations on subtypes vary somewhat, it is still true that only symptomatic disease should be treated. The definition of symptomatic disease has remained largely unchanged over the past 30 years and includes local symptoms due to lymphoma growth, impairment of normal organ function (e.g., symptomatic anemia), B symptoms, symptomatic extranodal disease, other cytopenias, or a rapid growth rate of a lymphoma manifestation. These criteria are very elastic and provide the patient and the physician with a large decision framework. Therefore, efforts are currently underway to establish a prognostically better classification and thus decision support by means of new (molecular) markers. The recently presented CLL prognostic index (CLL-IPI), which provides a recommendation regarding the timing of initiation and type of therapy, serves as one example [3].
Radiotherapy in early stages: a curative option?
Radiotherapy has been defined in recent decades as a curative treatment option for early stages of follicular lymphoma. 15-25% of all patients diagnosed with follicular lymphoma present with stage I or II disease, and current national as well as international guidelines still recommend that radiotherapy be performed in these patients [4].
The difficulty of making a positive recommendation for the implementation of radiotherapy in today’s clinical practice is based on the uncertainty whether the data of old studies (large-area irradiation surfaces, high total doses, non-lymph node- or involuted-field-specific applications) are transferable to today’s irradiation techniques. Thus, the major dilemma is that we are currently unable to define the putative curative value of radiotherapy in early-stage indolent lymphoma (primarily follicular lymphoma).
Waiting after diagnosis: Is it still valid?
Some authors estimate that currently about 50% of patients still do not require immediate treatment at the time of diagnosis. The term “watch & wait” (w&w) was coined more than 35 years ago [5]. Waiting seemed reasonable when the disease progressed slowly and there were no or only mild symptoms – and also because no effective therapies existed. Thus, at that time, the median time to first therapy in FL patients was 31-36 months. Longitudinal observational analyses showed that approximately 20% of FL patients did not require therapy at a median follow-up of 17 years. Compared with a cohort of patients treated with chemotherapy at diagnosis, there was no difference in 5-year overall survival (OS). The median OS was eleven years and varied widely among histologies.
Whether w&w is appropriate affects all iNHL, but is analyzed primarily at the FL. Thus, in a multicenter study, 379 patients were randomized to three treatment arms: observation alone (w&w), rituximab induction alone (four-weekly applications), or rituximab induction followed by two years of maintenance therapy (rituximab every two months) [6]. This showed a significant benefit in progression-free survival (PFS) for both rituximab arms compared with w&w. However, overall survival did not differ, so even today there is no compelling reason for the early use of immunotherapy. This may change with new (immuno)therapeutics and should therefore always be questioned.
Current therapies
Treatment options for iNHL have changed dramatically in recent years with the establishment of new therapies. These are primarily based on antibodies and tyrosine kinase inhibition (Fig. 3) . So far, immunotherapy with CD20-specific antibodies as monotherapy or in combination with classical chemotherapeutic agents dominates first-line therapy. It must still be assumed that a cure is not possible despite modern therapeutic procedures; the only exception is allogeneic stem cell transplantation.
CD20-specific monoclonal antibodies
A classic example of a CD20-specific antibody is rituximab, which is now an integral part of B-cell lymphoma treatment and can be used for various entities either as monotherapy (e.g., FL) or in combination with chemotherapy (e.g., CLL). Newer anti-CD20 antibodies (e.g. obinutuzumab) have in some cases even higher direct cell toxicity (ADCC activity) and can thus eliminate lymphoma cells more efficiently [7]. In CLL treatment, for example, this leads to an increase in patients with negative minimal residual disease (MRD). This means that in patients treated with obinutuzumab + chemotherapy, the disease is more often undetectable (38 to 3% in peripheral blood) compared to conventional treatment with rituximab + chemotherapy, despite sensitive detection methods. It remains to be seen whether the increased rate of MRD negativity will also result in prolonged survival.
Combination of antibody and chemotherapy
Typically, CD20-specific antibodies are used in combination with chlorambucil, bendamustine, or CHOP, respectively. In this context, chlorambucil is often used in combination with obinutuzumab in the treatment of elderly CLL patients or rituximab in MALT lymphoma [8]. In contrast, bendamustine in combination with rituximab is considered the first-line standard in follicular lymphoma (grades 1 and 2) [9] and CLL (with reservations about fludarabine-endoxan). Rituximab with CHOP is used somewhat less frequently than in the past and is primarily used for blastoid lymphoma variants or FL grade 3 . In general, the number of chemotherapy cycles is often limited to six (every three to four weeks) and immunotherapy is given for the same length of time or still as monotherapy for two years in the sense of maintenance therapy.
Immunomodulators
Immunomodulators (IMID) are small molecules (“small molecules”) that are usually taken orally. IMIDs such as lenalidomide were first used successfully in multiple myeloma, and are now also being tested in lymphoma with encouraging response rates. The combination of lenalidomide and rituximab in patients with FL grade 3 and relapsed or refractory disease achieved an overall response rate (ORR) of up to 86%. When the combination is used directly in first-line therapy, ORRs of up to 98% can be achieved with high rates of complete remission (CR) (87% CR and unconfirmed CR) and MRD negativity. Recently, approval was also granted for the treatment of mantle cell lymphoma (MCL). Approval is based on studies with relapsed or refractory MCL disease due to an ORR of 42-53% [10,11].
Inhibition of the B-cell receptor (BCR) signaling pathway.
Even higher response rates in MCL treatment can be achieved by agents that inhibit the BCR downstream signaling pathway, e.g., Bruton tyrosine kinase (BTK) or PI3 kinase (PI3K) inhibitors [12]. Both kinases are frequently constitutively active in lymphoma cells and promote cell proliferation or survival.
BTK inhibitors
Ibrutinib is the first approved BTK inhibitor that irreversibly binds covalently to a cysteine residue (Cys-481) of tyrosine kinase, causing potent and sustained inhibition of enzymatic activity. In CLL, ibrutinib showed high activity. The approval is based on a comparative study with the CD20-specific monoclonal antibody ofatumumab (RESONATE study) in 391 pretreated CLL/SLL patients [13]. At a median follow-up of 9.4 months, ibrutinib (420 mg/d/po) significantly improved PFS and OS. At 12 months, OS was 90% in the ibrutinib group and 81% in the ofatumumab group. The overall response rate was significantly higher for ibrutinib (42.6 vs. 4.1%, p <0.001). Response rate and duration were independent of the presence of del17p or resistance to purine analogues. This demonstrates the high value of BTK inhibitors in this difficult-to-treat subgroup of CLL patients. The most common adverse events were diarrhea, fatigue, fever, and nausea.
The second entity approved in Switzerland concerns the treatment of relapsed MCL. The basis for approval was a multicenter, single-arm phase II study of 111 pretreated MCL patients at a dose of 560 mg ibrutinib once daily. The full publication reported an ORR of 66% with a CR rate of 17% and a median duration of response (DOR) of 17.5 months [4]. Interestingly, the response rate increased continuously during the course of treatment (so-called “incremental response under treatment”), so that – in contrast to classical chemotherapies – late remissions can also occur with continued therapy.
PI3K inhibitors
The PI3K family consists of a number of serine/threonine kinases that regulate growth, differentiation, metabolism, survival, and proliferation in various cells. For example, inhibition of the p110δ-unit leads to significant B-cell depletion and blockade of the BCR downstream signaling pathway. Therefore, most therapeutic approaches in lymphoma treatment focus on direct blockade of the p110δ-unit. The prototype of this substance class is idelalisib as a selective p110δ inhibitor.
Idelalisib was initially tested in a randomized phase III trial in 220 patients with relapsed CLL in combination with rituximab [15]. In the control arm, patients received rituximab plus placebo. With a median pretreatment of three agents, almost all cases had been preceded by rituximab and an alkylane or purine nucleotide analog. Nearly 40% of CLL patients also had the unfavorable genetic alteration of the p53 gene.
In terms of efficacy, there was a significantly higher response rate in the rituximab + idelalisib arm (81 vs. 13%), a significant prolongation of 1-year PFS (93 vs. 46%, p<0.001) and a significant prolongation of 1-year OS (92 vs. 80%, p=0.02). The superiority of the combination of rituximab + idelalisib was demonstrated for all subgroups.
Side effects that are significant for everyday clinical practice and that go beyond rituximab application are early or, in some cases, late-onset diarrhea. However, when comparing the side effect profile with other substances that might be used in this situation, such as ofatumumab, alemtuzumab or conventional cytostatics, idelalisib certainly scores positively.
As a monotherapy agent, idelalisib shows high efficacy in iNHL in the setting of relapse therapy. The compound is approved for the treatment of FL patients with relapsed disease who have received two prior lines of therapy [16]. In the underlying single-arm phase II study of 125 iNHL patients resistant to rituximab and alkylans, idelalisib was dosed at 150 mg twice daily until disease progression. The median time to response was 1.9 months, the median duration of response was 12.5 months, and the median PFS was 11 months. The most common grade 3 or higher adverse events were neutropenia (27% of patients), elevation of aminotransferases (13%), diarrhea (13%), and pneumonia (7%).
BCL-2 inhibitors
The anti-apoptotic BCL-2 protein is overexpressed in lymphoma cells and contributes to chemotherapy resistance. Selective, oral BCL-2 inhibitors such as venetoclax stop the proliferation of lymphoma cells and thus lead to tumor remissions in preclinical models. In an initial clinical trial of 106 patients with MCL (n=28), follicular lymphoma (n=29), diffuse large B-cell lymphoma (n=41), and other NHL subtypes (n=8), venetoclax monotherapy demonstrated an acceptable safety profile at the maximum tolerated dose of 1200 mg/d [17]. The ORR was 44% for all subtypes, 78% for MCL, and 38% for FL. The median PFS was 10-14 months. The most common treatment-related adverse events (AEs ≥20%) were nausea (48%), diarrhea (44%), fatigue (41%), decreased appetite (21%), and vomiting (21%). Of importance is the occurrence of tumor lysis syndrome, which, however, occurred in two patients without clinical consequences.
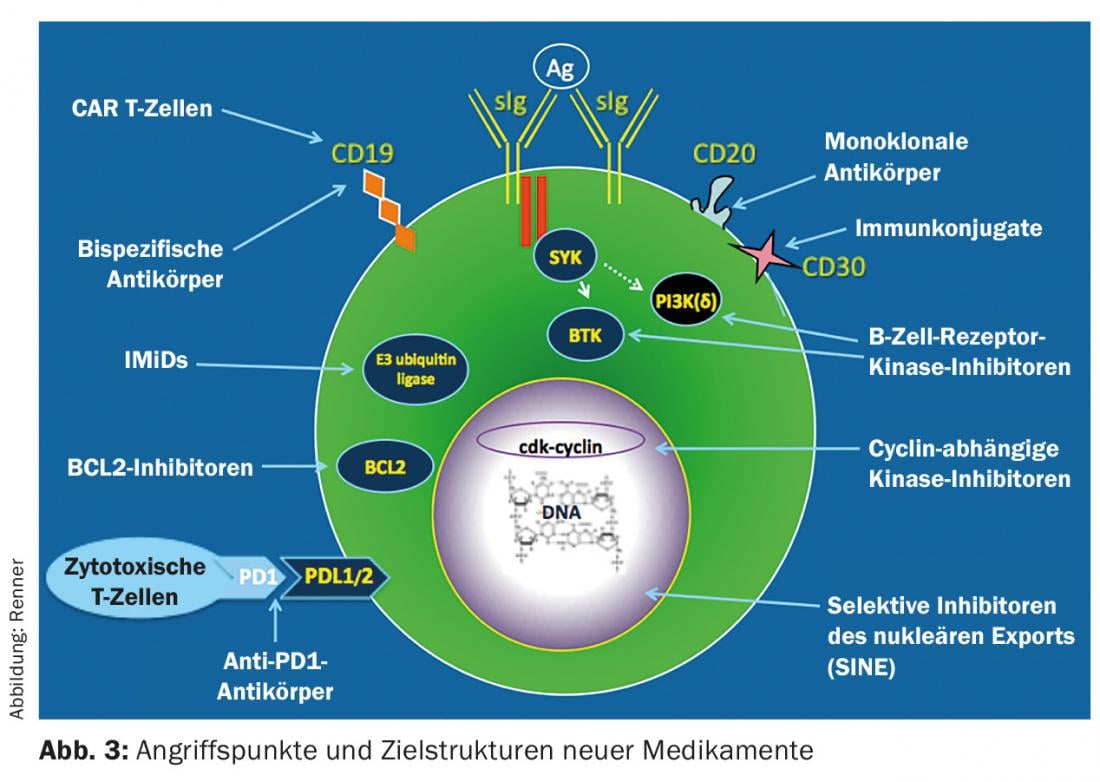
Future therapies
Future therapies will come primarily from two areas (Fig. 3): Signaling pathway inhibitors that target key switching molecules of the lymphoma cell and block their function (e.g., CDK 4/6 inhibitors), and novel immunotherapeutics. The recognition that the immune system contributes to tumor control is currently revolutionizing hemato-oncology treatment options. Checkpoint blockade inhibitors, bispecific antibodies and reprogrammed T cells (CAR T cells) are in clinical development with some impressive results. Perhaps one day we will succeed in stimulating the immune system in such a way that long-term tumor control and even cure will be possible.
Literature:
- Swerdlow SH, et al: IARC Press, Lyon, 2008.
- Brice P, et al: J Clin Oncol 1997; 15(3): 1110-1117.
- Molica S, et al: Abstract 498, presented at the 57th Amercan Society of Hematology (ASH) Annual Meeting, 2015.
- Hiddemann W, et al: Internist (Berl) 2016; 57(3): 222-229.
- Morrison VA, Peterson BA: Leuk Lymphoma 1993; 10 Sup: 29-33.
- Adreshna KM, et al: Lancet Oncol 2014; 15(4): 424-435.
- Goede V, et al: NEJM 2014; 370(12): 1101-1110.
- Zucca E, et al: J Clin Oncol 2013; 31(5): 565-572.
- Rummel MJ, et al: Lancet 2013; 381(9873): 1203-1210.
- Habermann TM, et al: Br J Haematol 2009; 145(3): 344-349.
- Witzig TE, et al: Ann Oncol 2011; 22(7): 1622-1627.
- Mato A, et al: Am J Hematol 2015; 90(7): 657-664.
- Byrd JC, et al: NEJM 2014; 371(3): 213-223.
- Wang ML, et al: NEJM 2013; 369(6): 507-516.
- Furman RR, et al: NEJM 2014; 370(11): 997-1007.
- Gopal AK, et al: NEJM 2014; 370(11): 1008-1018.
- Roberts AW, et al: NEJM 2016; 374(4): 311-322.
InFo ONCOLOGY & HEMATOLOGY 2016; 4(2): 11-15.