
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a rare ion channel disease without macroscopically detectable structural cardiopathy. It occurs exclusively in childhood and adolescence and has a high mortality if untreated. Therefore, therapy of the structurally unremarkable heart is of particular importance.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a rare ion channel disease without macroscopically detectable structural cardiopathy with a prevalence of 1:5000-10,000. First described in 1975 [1], characterization as a separate entity did not occur until 1995 [2]. Characteristic is the occurrence of polymorphic ventricular extrasystoles and ventricular tachycardia (VT) as well as bidirectional VT under physical or mental stress with an unremarkable resting ECG. Although isolated case reports of symptomatic initial manifestation exist up to 40 years of age, it is predominantly a disease of children and adolescents. Depending on the genetic subtype, the initial manifestation occurs between the ages of 2 and 20 with a familial cluster in 30% of cases. Men and women are equally affected, with earlier presentation in men [3]. Family screening should always be performed [4].
Pathophysiology
Although great progress has been made in characterizing the underlying disease mechanisms since the initial description, they are still not fully understood.
Defects in diastolic calcium (Ca2+) release from the sarcoplasmic reticulum (SR) result in Ca2+ overload in the myocardial cell. In healthy individuals, extracellular Ca2+ influx via L-type channels triggered by the action potential results in Ca2+-triggered Ca2+ release from the SR by activation of the ryanodine receptor (RYR2). This leads to myocardial contraction and plateau phase of the action potential with subsequent return of Ca2+ to the extracellular space and the SR. However, in CPVT, spontaneous diastolic Ca2+ release from the SR occurs in part because of an autosomal dominant defect in RYR2. This leads to a reversal of Ca2+ transport into the cell and to late afterdepolarizations, which form the basis for CPVT. If β-adrenergic stimulation by stress now occurs in addition, the characteristic polymorphic ventricular extrasystoles or tachycardias develop [5].
In addition to mutations in the RYR2 gene, which are detectable in approximately 50-55% of CPVT patients, there are other mutations of cardiac calsequestrin (CASQ2) and triadin (TRDN), two other components of the cellular Ca2+ budget [5]. In addition, dysfunction of potassium channels (KCNJ2) can also lead to CPVT. Depending on the detected genetic defect, one can distinguish between several subtypes. Overall, however, responsible genetic defects can be detected in only about 60% of affected individuals [5].
Diagnosis
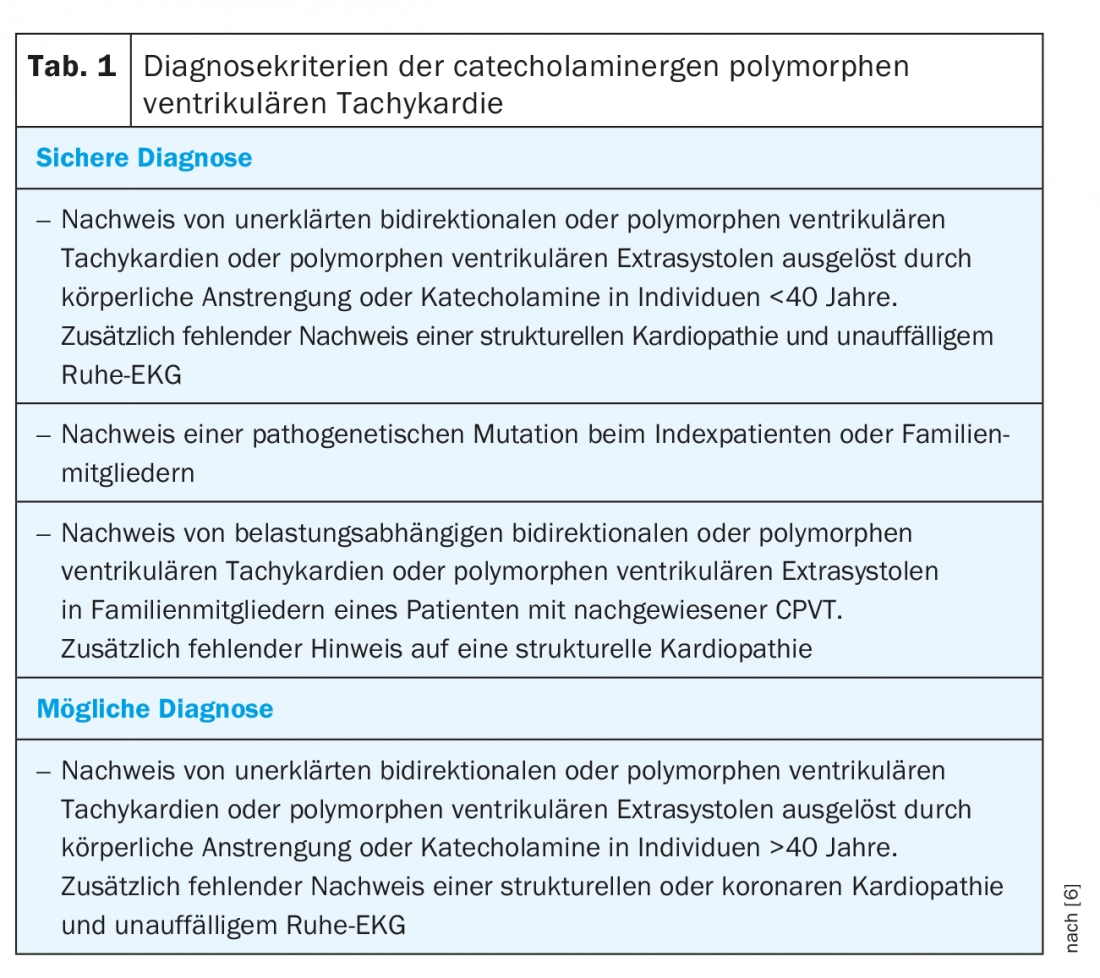
The diagnostic criteria of the professional societies are summarized in Table 1 [6]. Much more difficult than applying the diagnostic criteria, however, is identifying potential sufferers. Typical clinical presentations are children between 2-20 years of age with endured syncope or survived sudden cardiac death (SCD) during physical or emotional stress [2]. Because CPVT-related syncope can also cause convulsions and incontinence, children are often mistakenly treated for epilepsy. CPVT is then diagnosed late in the absence of efficacy of antiepileptic drugs. Similarly, a familial cluster of exercise-induced syncope or SCD and refractory familial epilepsy should suggest possible familial CPVT. Atypical presentation during cardiovascular screening by ergometry is possible in adulthood, but much less common.
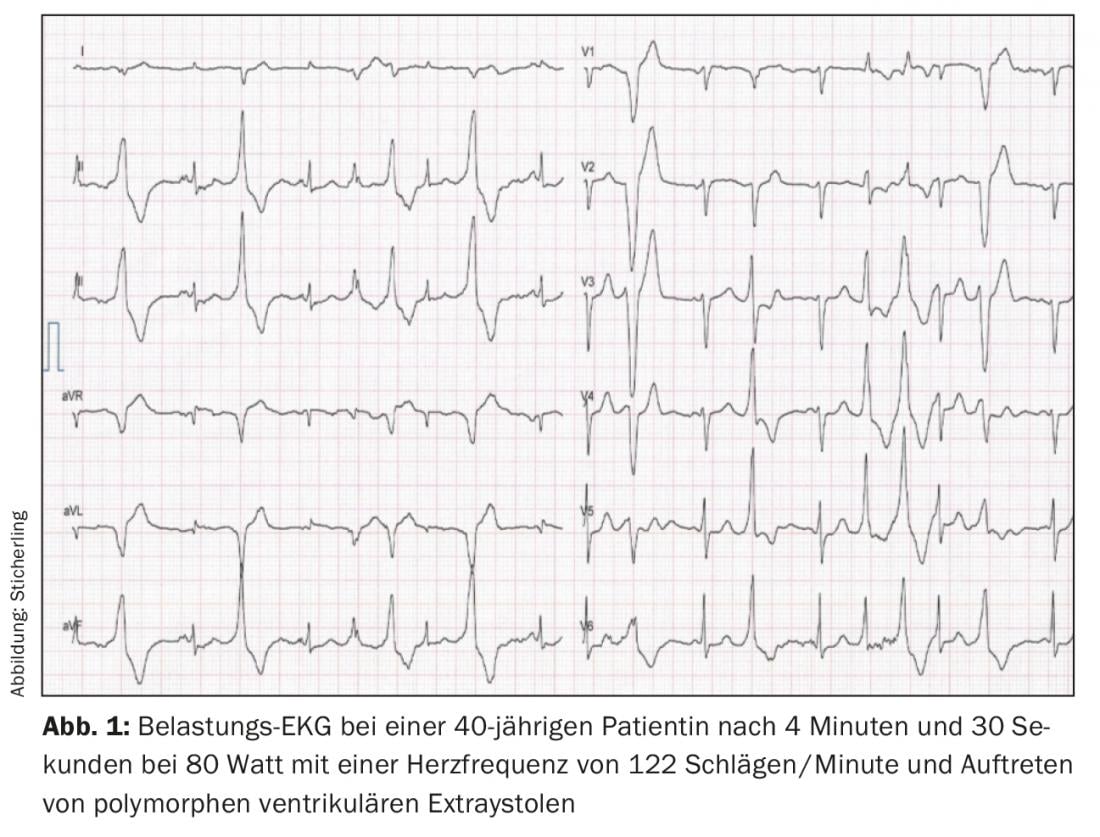
The gold standard for diagnosis is exercise ergometry. Typically, an individually reproducible heart rate threshold between 110-130 beats/minute can be identified, above which the first isolated ventricular extrasystoles occur with a coupling interval of approximately 400 ms. The extrasystoles predominantly show a superior axis with left bundle branch block or an inferior axis with right bundle branch block [4,7]. With increasing stress and heart rate increase, more frequent monomorphic extrasystoles occur, followed by bigeminy and finally polymorphic extrasystoles and/or non-sustained VT (Fig. 1). Rarely, polymorphic sustained VT or ventricular fibrillation may occur. The stress test should therefore be stopped if arrhythmogenic symptoms increase or the duration of nonsustained VT increases [7,8]. Long-term ambulatory ECG may have additional diagnostic utility in very young patients, in patients unable to perform exercise ECG, or in patients with negative exercise ECG but continued suspicion of CPVT. However, the sensitivity is lower than that of exercise ECG [9]. Another diagnostic option is catecholamine infusion. However, compared to the stress test, there is a lower sensitivity of about 75%, so it should be used for diagnostic purposes only in exceptional cases [10]. Electrophysiological examination with programmed stimulation has no value in the diagnosis of CPVT [2,10]. As mentioned above, resting ECG and imaging techniques do not provide diagnostic utility. However, normal findings in these examinations are mandatory to make the diagnosis [6].
Differentially, other ion channel diseases such as long QT syndrome must be considered, especially in SCD during physical activity such as swimming [11]. An exercise ECG can unmask QTc prolongation in the recovery phase that is undetectable on the resting ECG, helping to differentiate it from CPVT [12]. Another differential diagnosis is Andersen-Tawil syndrome, which, like a subtype of CPVT, is associated with a mutation in KCNJ2 and may also present with bidirectional ventricular tachycardia [13]. In addition to the phenotypic signs of periodic paralysis and dysmorphia of extremities that are not always present, exercise ECG may also help to differentiate this from CPVT. Differentiation is important because patients with Andersen-Tawil syndrome have a more benign prognosis [13]. In addition to ion channel diseases, one should always consider as yet undiagnosed structural diseases such as arrhythmogenic, hypertrophic, ischemic, or valvular cardiomyopathies, which are much more common overall. Other causes of bidirectional VT include digoxin intoxication or myocarditis [14,15].
Therapy
Treatment of CPVT consists of lifestyle modifications, drug therapy, risk stratification for an ICD to left cardiac sympathetic denervation (LKSD). Since mortality is up to 50% in severely affected patients, adequate therapy is of high importance [2,16] (Tab. 2) . In addition, familial screening should always be performed due to the autosomal dominant inheritance of the RYR2 defect.
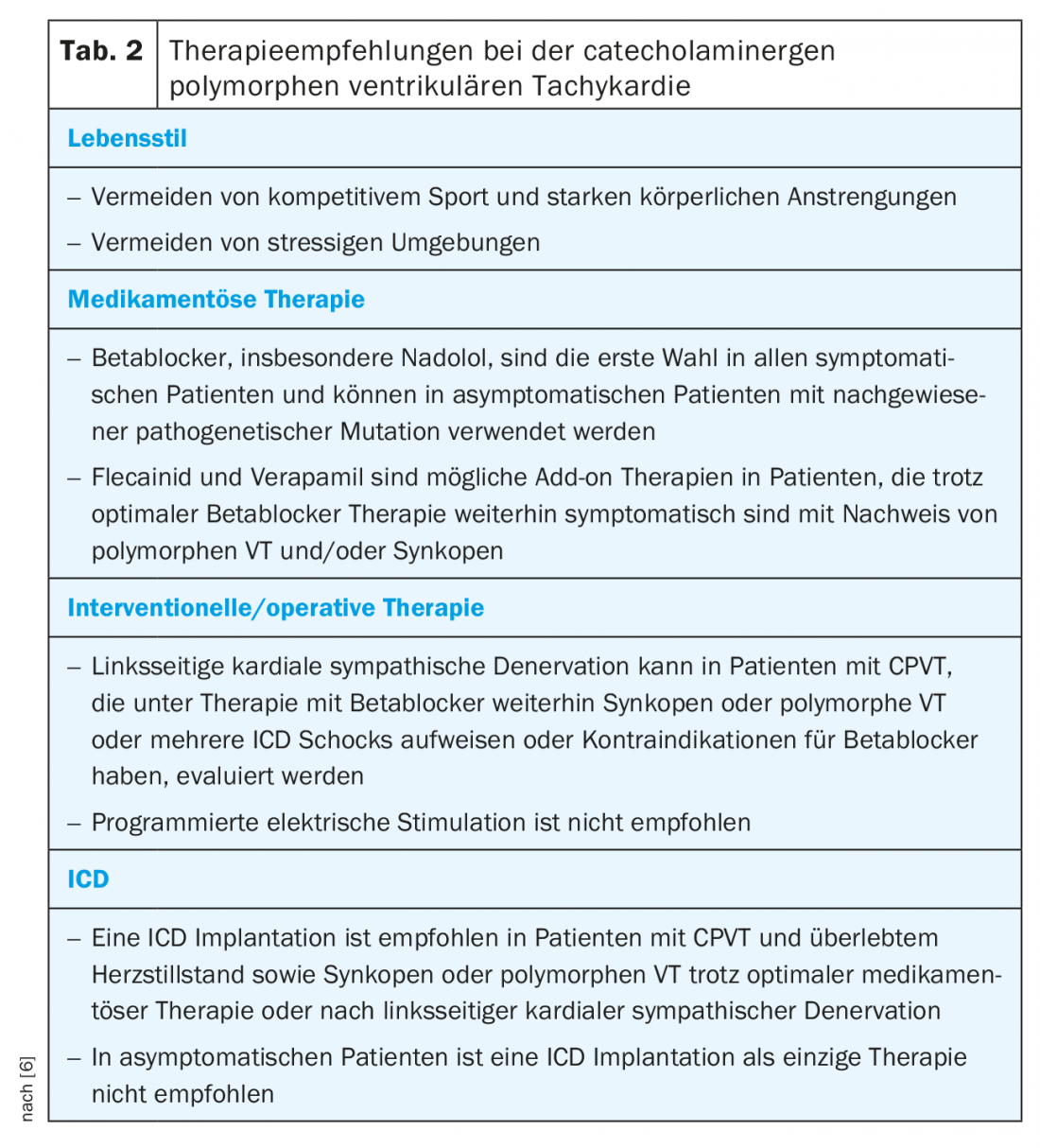
Because physical exertion can trigger ventricular arrhythmias, patients should avoid competitive exercise. Although a small study was able to show that in adequately treated CPVT patients, competitive exercise only resulted in more arrhythmias without an increase in mortality [17], transferability to all patients is not possible. Especially when swimming, a supervisor should be present.
Beta-blockers are the mainstay of drug therapy because, among other things, they prevent the arrhythmogenic heart rate limit from being reached. Most data are available for the long-acting, nonselective nadolol. However, this preparation is no longer available in many countries, such as Switzerland [16,18,19]. An alternative is carvedilol, which has a proven effect on RYR2 [20]. However, clinical data in CPVT patients are lacking. In principle, a complete dosage should always be taken up to the maximum tolerable dose. It is important to note that in children the dose must be adjusted to body weight. Adequate beta-blocker therapy can reduce the rate of fatal SCD to 6.4% over 8 years [19]. Similarly, genotype-positive patients who are detected by family screening but do not show arrhythmias on exercise ECG should receive beta-blocker therapy [6]. If arrhythmias or couplets and clustered ventricular extrasystoles continue to be seen on exercise ergometry with adequate beta-blocker therapy, additional therapy with flecainide should be evaluated. Flecainide likewise exhibits a direct inhibitory effect on RYR2 and has also been shown in clinical trials to suppress ventricular arrhythmias in 76% of patients already receiving beta-blocker therapy [21,22]. Calcium channel blockers appear to have additional benefit as an alternative add-on therapy. In small, nonrandomized studies, verapamil in addition to beta-blockers has been shown to further reduce the incidence of ventricular arrhythmias [23,24]. Larger randomized studies on verapamil are currently not available. Therapeutic options not yet used in clinical practice but potentially promising include propafenone, which showed promise in limited patient numbers [25] and dantrolene, which showed effectiveness in a cell model [26].
If medical therapy does not show adequate control of arrhythmias, an LCSD can be evaluated, in which the lower half of the left stellate ganglion and thoracic ganglia T2-4 are removed thoracoscopically. This results in a marked decrease in noradrenaline secretion in the heart with very good results in small series of patients with severe CPVT. The most common complication of LKSD is a mostly transient Horner’s syndrome, but this can also persist in 2-3%. Less common complications include injury to surrounding structures such as the pleura, phrenic nerve, brachial plexus, and somatic nerves with resultant shooting pain in the left shoulder. As a side effect, there may be a lack of sweating of the left hand and left forehead with warmer and drier skin compared to the side [27,28].
ICD implantation should be performed only in selected patients who have survived an SCD or continue to experience syncope or polymorphic or bidirectional VT despite optimal therapy. If LKSD is available, it should also be performed before implantation [6]. This restrictive recommendation exists primarily because ICD shocks per se lead to additional release of catecholamines. These can then trigger further arrhythmias and lead to a vicious circle, so the ICD should be programmed with high cut-offs and long delays before shock delivery [6,29].
Summary
CPVT is an extremely rare disease that occurs almost exclusively in children and adolescents and has a high mortality if left untreated. Beta-blocker therapy, along with lifestyle adjustment, is the most prognostically important intervention. ICD implantation is associated with specific, potentially proarrhythmic effects with possible shock delivery, and the indication in primary prophylaxis should therefore be reserved. Further studies are needed to better understand the disease pattern and to find new therapeutic approaches.
Take-Home Messages
- CPVT occurs almost exclusively in childhood and adolescence in structurally unremarkable hearts and has a high mortality if untreated.
- The main therapies are lifestyle adjustment and beta-blockers.
- ICD implantation should be performed only in patients with survived sudden cardiac death, syncope, or persistent polymorphic ventricular tachycardia despite maximal drug therapy or after evaluation of left-sided cardiac sympathetic denervation.
- Shock delivery by an ICD has a high risk of proarrhythmia, so long detection times and high intervention frequencies should be programmed into the ICD.
Literature:
- Reid DS, et al: Bidirectional tachycardia in a child. A study using His bundle electrography. Br Heart J. 1975; 37:339-344.
- Leenhardt A, et al: Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995; 91: 1512-1519.
- Priori SG, et al: Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002; 106: 69-74.
- Leenhardt Antoine, Denjoy Isabelle, Guicheney Pascale. Catecholaminergic Polymorphic Ventricular Tachycardia. Circ Arrhythm Electrophysiol. 2012; 5: 1044-1052.
- Pérez-Riera AR, et al: Catecholaminergic polymorphic ventricular tachycardia, an update. Ann Noninvasive Electrocardiol Off J Int Soc Holter Noninvasive Electrocardiol Inc 2018;23: e12512.
- HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes. 2013; 69.
- van der Werf C, Wilde AAM: Catecholaminergic polymorphic ventricular tachycardia: from bench to bedside. Heart. 2013; 99: 497-504.
- Svendsen JH, Geelen P, EHRA Scientific Initiative Committee: Screening for, and management of, possible arrhythmogenic syndromes (channelopathies/ion channel diseases). Eur Eur Pacing Arrhythm Card Electrophysiol J Work Groups Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2010; 12: 741-742.
- Sy RW, et al: Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2011; 8:864-871.
- Sumitomo N, et al: Catecholaminergic polymorphic ventricular tachycardia: electrocardiographic characteristics and optimal therapeutic strategies to prevent sudden death. Heart. 2003;89: 66-70.
- Tester DJ, et al: Unexplained drownings and the cardiac channelopathies: a molecular autopsy series. Mayo Clin Proc. 2011; 86: 941-947.
- Sy RW, et al: Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation. 2011; 124: 2187-2194.
- Inoue YY, et al: Different responses to exercise between Andersen-Tawil syndrome and catecholaminergic polymorphic ventricular tachycardia. Eur Pacing Arrhythm Card Electrophysiol J Work Groups Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2018; 20: 1675-1682.
- Berte B, et al: Bidirectional ventricular tachycardia in fulminant myocarditis. Eur Pacing Arrhythm Card Electrophysiol J Work Groups Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2008; 10: 767-768.
- Valent S, Kelly P: Images in clinical medicine. Digoxin-induced bidirectional ventricular tachycardia. N Engl J Med 1997; 336: 550.
- Hayashi M, et al: Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009; 119: 2426-2434.
- Ostby SA, et al: Competitive Sports Participation in Patients With Catecholaminergic Polymorphic Ventricular Tachycardia: A Single Center’s Early Experience. JACC Clin Electrophysiol. 2016; 2: 253-262.
- Leren IS, et al: Nadolol decreases the incidence and severity of ventricular arrhythmias during exercise stress testing compared with β1-selective β-blockers in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2016; 13: 433-440.
- van der Werf C, Zwinderman AH, Wilde AAM: Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: state of the art and future developments. Eur Eur Pacing Arrhythm Card Electrophysiol J Work Groups Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2012; 14:175-183.
- Zhou Q, et al: Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med 2011; 17: 1003-1009.
- van der Werf C, et al: Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. 2011; 57: 2244-2254.
- Watanabe H, et al: Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 2009; 15: 380-383.
- Rosso R, et al: Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007; 4: 1149-1154.
- Swan H, et al: Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J Cardiovasc Electrophysiol. 2005; 16: 162-166.
- Hwang HS, et al: Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2011; 4: 128-135.
- Jung CB, et al: Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012; 4: 180-191.
- Odero A, et al: Left cardiac sympathetic denervation for the prevention of life-threatening arrhythmias: the surgical supraclavicular approach to cervicothoracic sympathectomy. Heart Rhythm. 2010; 7: 1161-1165.
- Wilde AAM, et al: Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008; 358: 2024-2029.
- Mohamed U, et al: Sudden cardiac death despite an implantable cardioverter-defibrillator in a young female with catecholaminergic ventricular tachycardia. Heart Rhythm. 2006; 3: 1486-1489.
CARDIOVASC 2019; 18(2): 12-15









