Loss of function of the cystic fibrosis transmembrane conductance regulator (CFTR) channel results in the formation of a highly viscous secretion in cystic fibrosis, leading to a decrease in mucociliary clearance. Patients report chronic cough and purulent sputum and increased respiratory infections. Atelectasis may occur due to secretion obstruction; further complications may include pneumothoraces or hemoptysis.
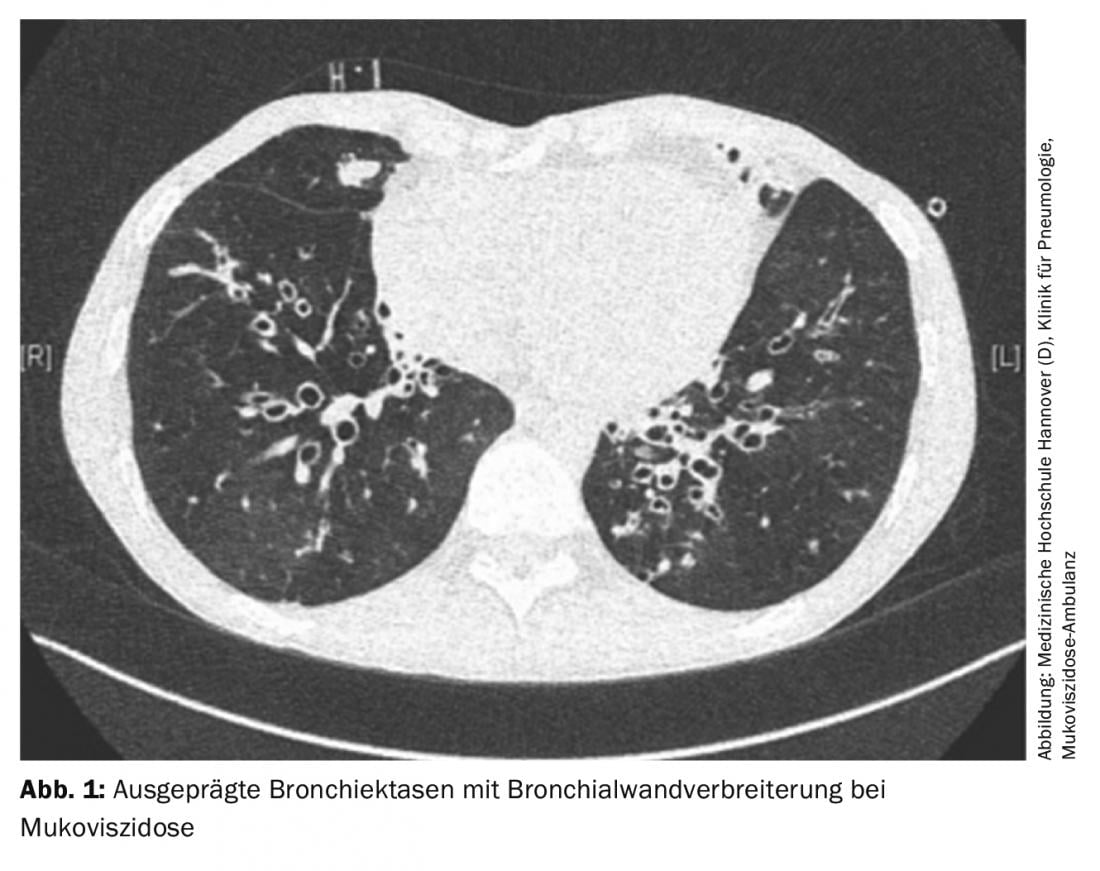
Loss of function of the cystic fibrosis transmembrane conductance regulator (CFTR) channel results in the formation of a highly viscous secretion in cystic fibrosis, leading to a decrease in mucociliary clearance. This favors bacterial colonization and leads to inflammation and destruction of lung tissue with subsequent respiratory insufficiency. Lung functionally, progression of the disease usually reveals obstructive ventilation disorder with hyperinflation. Patients report chronic cough and purulent sputum and increased respiratory infections. Imaging often shows mucus-filled bronchiectasis with thickened bronchial walls (Fig. 1). Atelectasis may occur due to secretion obstruction; further complications may include pneumothoraces or hemoptysis.
According to these symptoms, the treatment of lung disease focuses on secretolysis, antibacterial and anti-inflammatory therapy.
Secretolysis
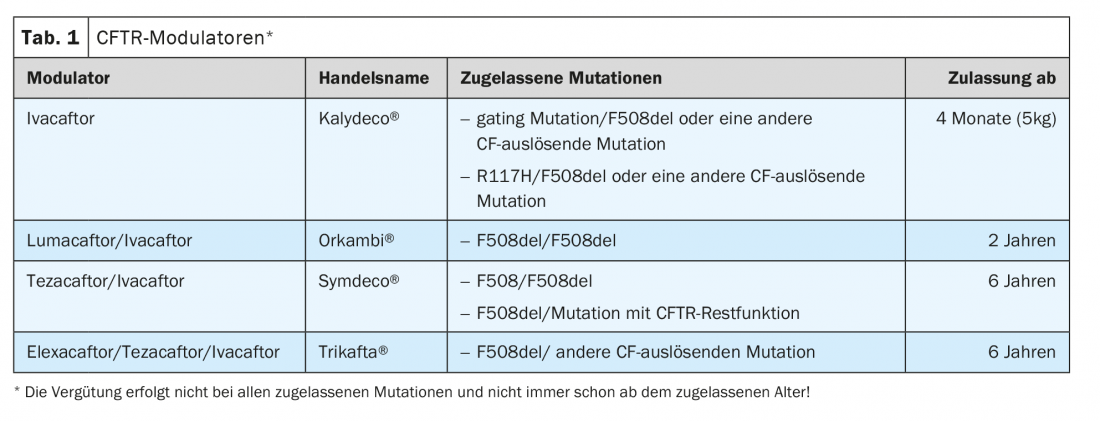
CFTR modulators: The CFTR channel is an anion channel for chloride and bicarbonate. This membrane-bound protein is localized in the respiratory epithelium but also in the gastrointestinal tract and many other organs. The first CFTR modulator, ivacaftor, was approved 10 years ago as the first therapy to affect the underlying base defect of cystic fibrosis (Table 1).
Ivacaftor
CFTR modulators are distinguished between potentiators and correctors. Potentiators, of which ivacaftor is one, increase the opening probability of the CFTR channel by activating ATP-independent mechanisms. Accordingly, clinical studies showed an effect in mutations where the CFTR ion channel is present in the cell membrane but the opening probability needs to be increased. FEV1 increased by 10.6% points in patients with at least one G551D mutation (c.1652G>A). The likelihood of having a pulmonary exacerbation decreased by 55%, and quality of life and weight increased.
These results led to the approval of ivacaftor in 2012 for gating mutation, but these are rare, so about 7% of CF patients could receive the drug. Registry studies from the UK and the USA show that the positive effects are still detectable 4 and 5 years after the start of therapy, respectively.
Lumacaftor
In about 50% of CF patients, the F508del mutation is present in homozygous form. In this very large group, ivacaftor therapy had not proven effective. Phase 2 studies showed evidence that a combination of ivacaftor with lumacaftor could result in clinical improvement. Lumacaftor belongs to the CFTR correctors. It increases the amount of CFTR protein in the cell membrane by changing the processing in the cell.
Phase 3 studies showed a significant effect of this combination therapy, although it was smaller than the effect of ivacaftor on gating mutations. FEV1 increased by 3% points, and exacerbations decreased by 30%. This therapy was approved for this patient group in 2016.
Tezacaftor
Two years later, data on another CFTR modulator, the corrector tezacaftor in combination with ivacaftor, were published. In a placebo-controlled phase 3 study in patients with homozygous F508del mutation, the combination treatment of tezacaftor and ivacaftor showed superiority with a 4%-point increase in FEV1. Again, the rate of pulmonary exacerbations was 35% lower than in the placebo group. Thus, the effect was comparable to that of Orkambi in F508del homozygous patients.
Another study compared efficacy in patients who had F508del and another CFTR mutation with residual function. These patients were divided into three groups, with one group receiving tezacaftor in combination with ivacaftor, the second group receiving ivacaftor alone, and the third group receiving placebo. Both treatment groups were superior to the placebo group in terms of FEV1. Ivacaftor alone resulted in an increase of 4.7% points, while the combination treatment even resulted in an increase of 6.8% points.
Based on these results, the combination treatment was approved for both F508del homozygous patients and patients who have F508del and another CFTR mutation with residual function.
Elexacaftor/Tezacaftor/Ivacaftor
Another major step in improving therapy is evident in the clinical studies on the efficacy of the triple combination of elexacaftor, tezacaftor and ivacaftor. In a placebo-controlled study published in 2019, pa-tients who had an F508del and another CFTR mutation with minimal function (approximately 200 different mutations) were studied.
The results of this study significantly exceeded previous findings. Lung function showed an increase in FEV1 after only 2 weeks of therapy, which remained stable at 13.6% points until week 24, and the rate of pulmonary exacerbations was reduced by 60%. The patients’ quality of life increased significantly (18.1 points in the CFQ-R), as did their weight (BMI +1.3). Welding tests were performed to assess CFTR function during therapy. There was a marked decrease of 40 mmol/l in the sweat chloride concentration pathologically elevated in cystic fibrosis, so that many patients were no longer above the 60 mmol/l limit required for the diagnosis of cystic fibrosis.
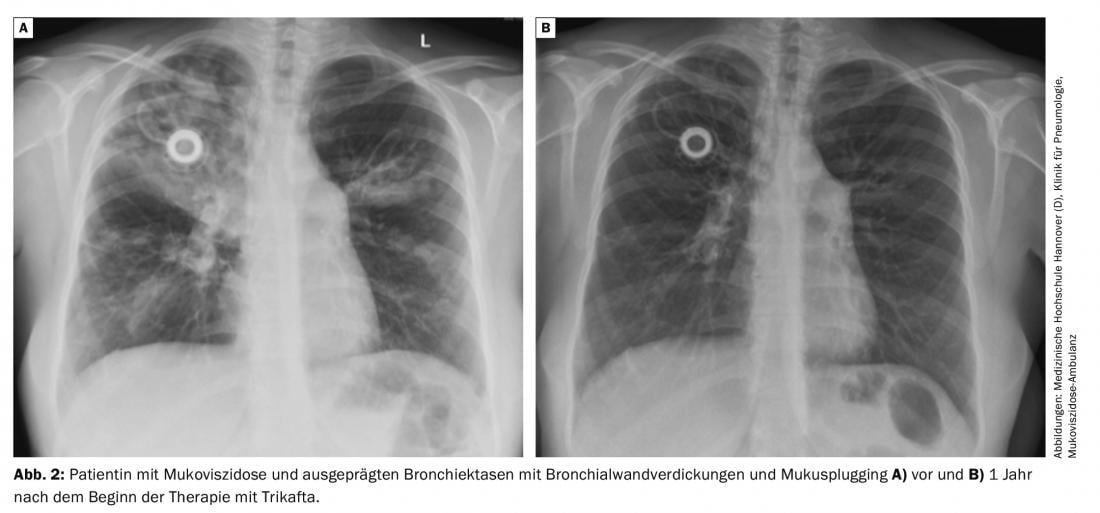
The efficacy of the triple combination was additionally tested in F508del homozygous patients. Tezacaftor and ivacaftor therapy served as controls in this study, which lasted only 4 weeks. The triple combination was clearly superior to therapy with tezacaftor and ivacaftor, with a 10%-point increase in FEV1, and there were also significant benefits in quality of life and chloride content of the sweat test (Fig. 2) .
Therapy with elexacaftor/tezacaftor/ivacaftor is usually well tolerated. Adverse events that occurred more frequently in the verum group than in the placebo group during the clinical trial were skin rashes (10%), increased liver enzymes (10%), headache, and diarrhea. 1% of patients discontinued therapy due to adverse events.
CFTR modulators must be taken with a meal containing fat. Patients must not eat grapefruit or bitter oranges or take St. John’s wort because of interactions; dosage must be adjusted for concomitant use of moderate or strong CYP3A inhibitors (e.g., azole antifungals, antidepressants).
The cost of therapy with CFTR modulators is CHF 12,000-17,000 per month. Therefore, the indication, initiation, and monitoring of therapy must be performed by a center with experience in the treatment of cystic fibrosis.
Behind these many impressive figures lies a fundamental change in the quality and perspective of life in everyday life. Many patients no longer have a cough or sputum, symptoms that were previously constant companions. Although there are no data yet on the long-term course, it can be assumed that life expectancy is significantly increased by these drugs.
Nevertheless, the medication does not work equally in all patients, and patients with severely destroyed lungs continue to have impaired lung function. Therapy is not approved for approximately 10% of patients and approximately 1-2% of patients discontinue therapy due to intolerance. Therefore, other forms of therapy continue to be important.
Dornase alfa: In the context of respiratory tract inflammation, DNA is released by the disintegration of granulocytes. Dornase alfa cleaves DNA and thereby reduces the viscosity of the secretion. In clinical trials, this resulted in a reduction in pulmonary exacerbations and improvement in lung function.
Hypertonic saline and mannitol: Due to an osmotic effect, there is a reduction in sputum viscosity after inhalation of hypertonic saline or mannitol, resulting in improved mucociliary clearance. To avoid airway obstruction, a bronchodilator should be used before inhalation.
Physical therapy and sports: Physical therapy and sports are part of the basic therapy, even though there are insufficient clinical studies to prove their effectiveness. Through therapy, patients learn techniques to improve mucociliary clearance, optimize coughing and inhalation technique, and perform thoracic mobilization and strength and endurance training.
Antibiotic therapies
Staphylococcus aureus: In children and adolescents, Staphylococcus aureus is the germ most frequently detected in bronchial secretions. There is no single strategy for treating this germ. There is evidence that there is increased inflammation of the respiratory tract by this germ and thus it has a role in the course of the disease. An eradication attempt with oral antibiotics is therefore frequently performed, particularly in children, even in the case of asymptomatic detection. In case of symptomatic detection, oral antibiotic therapy for 2-3 weeks should be given in any case.
With increasing age, chronic evidence of Staphylococcus aureus often becomes apparent. However, continuous staphylococcal therapy should be considered only in exceptional cases, e.g., frequent pulmonary exacerbations, because there is evidence that this therapy leads to increased evidence of Pseudomonas.
Pseudomonas aeruginosa: With increasing age, the frequency of detection of Pseudomonas aeruginosa and other gram-negative germs in bronchial secretions increases. Approximately 30% of 18-year-old and 75% of 45-49-year-old patients are chronically colonized with this germ. Since not every new germ detection is accompanied by clinical deterioration, pharyngeal swabs or sputum samples must be routinely examined approximately 4 times/year. Detection of Pseudomonas aeruginosa requires a long incubation period, so it must be ensured that the microbiology laboratory meets the appropriate requirements.
At the first detection of Pseudomonas aeruginosa, inhaled antibiotic therapy for eradication is indicated regardless of clinical symptoms. Different, equivalent protocols are available for this purpose:
- Tobramycin 2× 300 mg for 4 weeks
- Colistin 2× 1 million IU for 3 months in combination with ciprofloxacin 750 mg 2× 1 for 3 weeks.
- Aztreonam lysate 3× 75 mg for 4 weeks.
Eradication is considered successful if 6 subsequent controls are negative. This succeeds in about 80% of cases.
Chronic Pseudomonas aeruginosa colonization is said to occur when more than half of 6 controls in 12 months are positive. In this case, permanent antibiotic inhalation therapy is recommended as suppressive treatment. Inhalation results in higher sputum levels than i.v. administration and has lower toxicity. Bronchoconstriction after inhalation can be counteracted by bronchodilators. Powder inhalations are also available. While inhalation is more likely to cause coughing than moist inhalation, the reduced inhalation time is still a distinct advantage for many patients.
The following inhaled antibiotics are available:
Powder inhalation:
- Colistin 2× 1 hard capsule a 125 mg continuously
- Tobramycin 2× 4 hard capsules of 28 mg 28 days on/off
Moist inhalation:
- Aztreonam lysate 3× 75 mg. 28 days on/off
- Colistin 2× 1(-2) million IU continuous.
- Levofloxacin 2× 240 mg 28 days on/off (from 18 yr).
- Tobramycin 2× 80/160/170/300 mg continuous or 28 days on/off.
Pulmonary exacerbation
Time and again, patients experience pumonal exacerbations despite suppressive treatment. These are usually characterized by several of the following symptoms:
- Increased cough
- increasing dyspnea
- Change in sputum volume and color
- Fever
- Feeling of sickness, fatigue
- Weight loss
- Drop in FEV1 by >10%
- Increase in radiological changes
Pulmonary exacerbation can be treated by intensification of inhaled therapy, oral antibiotic therapy, or intravenous antibiotic therapy.
Ciprofloxacin and levofloxacin are available as oral, pseudomonas-active drugs. When prescribing, the risk of tendinitis to tendon rupture must be considered.
Intravenous therapy is usually administered as combination therapy over 14 days. The most commonly used drugs are ceftazidime, meropenem, and piperacillin/tazobactam combined with an aminoglycoside-usually tobramycin. Depending on the antibiogram, additional antibiotics may be used if resistance increases and standard therapies fail to work.
Anti-inflammatory therapy
Secretolytic measures and antibiotic therapies lead to a reduction in inflammation. Another anti-inflammatory therapy is treatment with azithromycin. The macrolide antibiotic, when used as continuous therapy, leads to an improvement in lung function and a reduction in exacerbations.
High-dose therapy with ibuprofen has shown beneficial effects in clinical trials but has not gained acceptance in clinical practice because of concerns about gastrointestinal or renal side effects.
Inhaled steroids may be part of therapy in patients with coexisting bronchial asthma. General use in cystic fibrosis is not indicated. The same is true for oral steroid therapy, which is used only in patients with allergic bronchopulmonary aspergillosis (ABPA).
Lung Transplant
For severely impaired lung function, lung transplantation represents an opportunity to improve quality of life and long-term prognosis. Presentation to the transplant center should be considered in the following cases:
- FEV1<30% or if there is a rapid decline in lung function.
- Frequent pulmonary exacerbations, esp. for intensive medical treatment
- recurrent pneumothoraces
- repeated hemoptysis
Internationally, 15% of lung transplants are performed for cystic fibrosis. It is likely that this number will decrease significantly due to the improved treatment options provided by CFTR modulators.
Polyposis nasi and chronic sinusitis
Many cystic fibrosis patients suffer from chronic sinusitis and polyposis nasi. The distress resulting from nasal obstruction, pain over the NNH, and decreased olfaction is often quite significant.
As a conservative, local therapy, nasal irrigation with NaCl 0.9% to remove secretions and crusts is the first priority. Inhalation devices, some of which produce additional vibration to achieve better deposition in the sinuses, can also be used. Topical steroids are particularly effective in allergy-associated nasal polyps, and a trial of therapy is appropriate in patients with cystic fibrosis, although neutrophilic inflammation is usually the primary concern. Topical α-sympathomimetics should be used infrequently because of tachyphylaxis.
The effect of CFTR modulators on polyposis nasi was not studied in the pivotal trials. Studies published in the meantime show that there is also a significant reduction in complaints in this area.
If there is no satisfactory improvement of the symptoms under conservative therapy, surgical therapy is possible. However, recurrence is very common after polyp removal alone.
Exocrine pancreatic insufficiency, recurrent pancreatitis
Exocrine pancreatic insufficiency is present in approximately 85% of CF patients. Resulting abdominal pain, bulky, foul-smelling stools with fatty deposits, and malnutrition represent an early leading symptom of the disease. Exocrine pancreatic insufficiency usually exists at birth. Therapy consists of substituting pancreatic enzymes at mealtime.
Recurrent pancreatitis occurs in approximately 20% of patients who do not have exocrine pancreatic insufficiency. These are often patients who have a more mild course in terms of pulmonary disease.
The therapy of pancreatitis is symptomatic. Anecdotal case reports suggest that CFTR modulators may have a beneficial effect on the incidence of pancreatitis. On the other hand, the occurrence of pancreatitis after the initiation of CFTR modulator therapy has been described.
Hepatobiliary complications
Approximately 40% of patients develop focal biliary fibrosis, which progresses to cirrhosis in approximately 20% of these patients. Therapeutic options are very limited. Ursodeoxycholic acid is recommended in European guidelines, but the data are weak. In individual cases, liver transplantation may become necessary. How CFTR modulators affect the development of fibrosis or cirrhosis is not yet known.
Malignancies
Patients with cystic fibrosis are at increased risk for gastrointestinal tumors. The performance of a colonoscopy as a preventive examination is therefore recommended from the age of 40.
Diabetes mellitus
The prevalence of diabetes mellitus in cystic fibrosis increases markedly with age, reaching approximately 30% by age 30.
Treatment is usually by insulin. A clinical trial showed that, at least in the first two years after diagnosis, oral therapy with repaglinide is not inferior to insulin therapy. The extent to which CFTR modulator therapy influences the development of diabetes mellitus cannot be conclusively assessed. Here, too, there are individual positive reports.
Male infertility
98% of men with CF are infertile due to prenatal congenital bilateral aplasia of the vas deferens (CBAVD) leading to azoospermia. Since sperm production is usually preserved, sperm can be obtained from the testis or epididymis and used for intrauterine insemination (IUI), in vitro fertilization (IVF), or intracytoplasmic sperm injection (ICSI).
Gynecological aspects in patients with CF
In patients with cystic fibrosis, there is only slightly reduced fertility, a.e. due to the increased viscosity of the cervical secretion. Numerous case reports suggest that normalization of viscosity by treatment with CFTR modulators leads to improvement in fertility.
Whether therapy with a CFTR modulator should be continued in the event of pregnancy must be determined on a case-by-case basis. The CFTR modulators cross the placental barrier, but adverse effects on the child have not been described. Animal studies also showed no evidence of harm.
Literature:
- Ramsey BW, et al: A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365(18): 1663-1672.
- Volkova N, et al: Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. JCF 2020; 10: 68-70.
- Wainwright CE, et al: Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med 2015; 373(3): 220-231.
- Taylor-Cousar JL, et al: Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med 2017; 377(21): 2013-2023.
- Rowe SM, et al: Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med 2017; 377(21): 2024-2035.
- Middleton et al. Elexacaftor-Tezacaftor-Ivacftor for Cystic Fibrosis with a Single Phe508del Allele. N Engl J Med 2019; 381(19): 1809-1819.
- Yang C, Montgomery M.: Dornase alfo for cystic fibrosis. Cochrane Database Syst Rev 2021; 3(3): CD001127; doi: 10.1002/14651858.CD001127.pub5.
- Wark P, McDonald VM: Nebulized hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev 2018; 9(9): CD001506; doi: 10.1002/14651858.CD001506.pub4.
- Beswick DM, et al: Impact of Cystic Fibrosis Transmembrane Conductance Regulator Therapy on Chronic Rhinosinusitis and Health Status: Deep Learning CT Analysis and Patient-reported Outcomes. Ann Am Thorac Soc 2022; 19(1): 12-19.
- Hewer SCL, et al: Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis: Cochrane Database Syst Rev 2017; 4(4): CD004197.
InFo PNEUMOLOGY & ALLERGOLOGY 2022; 4(4): 14-18.