
Autoimmune movement disorders occur very rarely and can appear like neurodegenerative diseases. That’s what makes it so difficult to make the right diagnosis. However, characteristic phenotypes and red flags may provide initial clues to the underlying antibodies.
Involuntary movements can have a variety of causes – and for this reason are not always causally treatable. Certain warning signs can be helpful for diagnosis. These include hemichorea, in which no structural changes were observed on MRI. Hyponatremia is also one of the red flags where antibodies should be considered, emphasized Prof. Bettina Ballint, MD, Zurich. LGI1 antibodies may manifest with hyponatremia and prodromal bradycardia. Before these patients develop severe cognitive deficits, they can be effectively treated. Faciobrachial dystonic seizures should also be monitored for LGI1 antibodies. If movement disorders also occur during sleep, IgLON5 antibodies could be causative. In addition, a strong stridor is conspicuous in these patients. IgLON5 antibodies operate in the gray area between autoimmunity and neurodegeneration. Patients exhibit both autoimmune susceptibility and, for example, hyperphosphorylated 3R and 4R tau in hypothalamus and tegmentum, without inflammatory infiltrates. The response of these patients to immunotherapy is very mixed.
IgLON5 tauopathy in neurodegenerative diseases in focus.
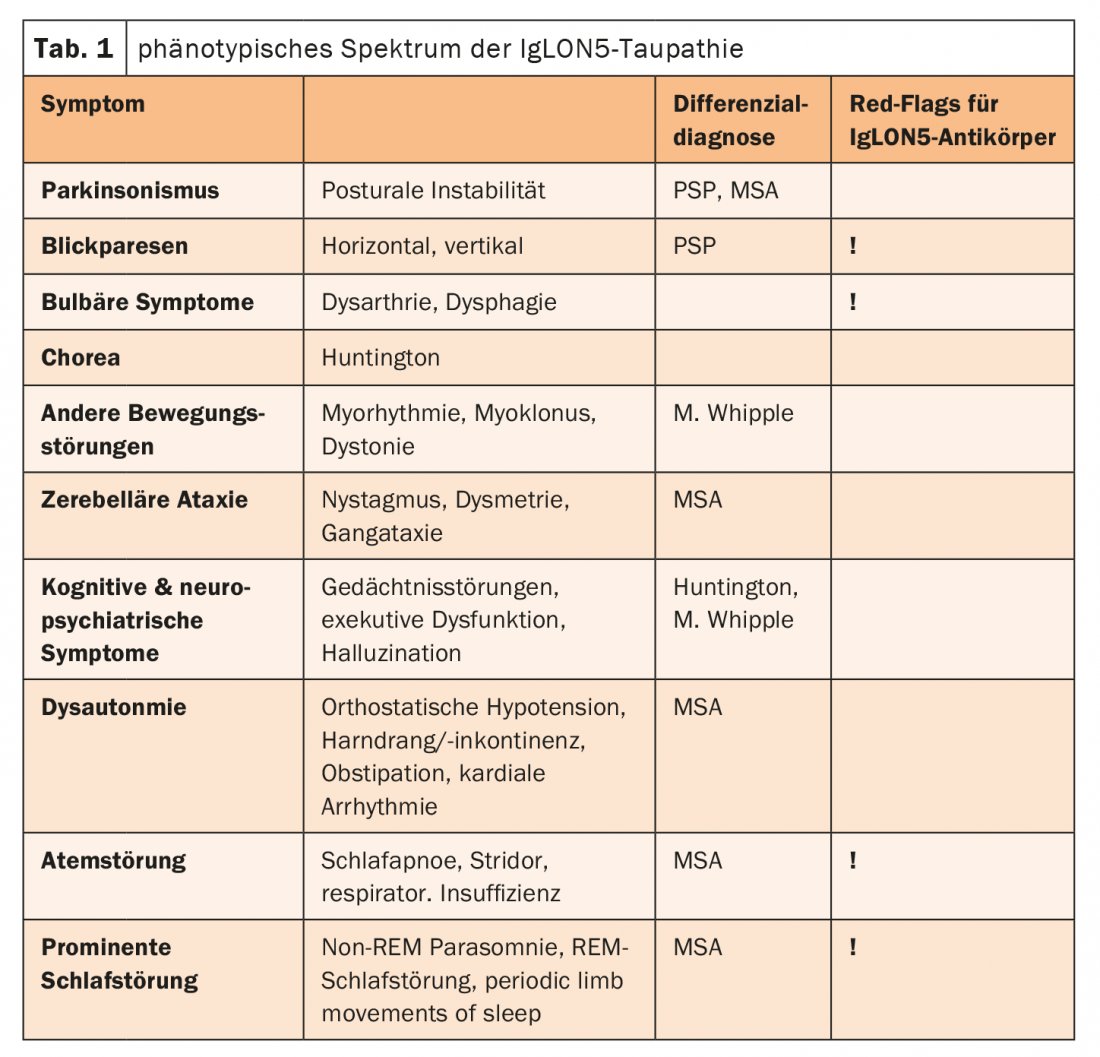
IgLON5 is a cell adhesion molecule on the surface of neurons and is relevant for neuronal pathfinding and formation of synapses as well as membrane stabilization. IgLON5-IgG1 antibodies lead to internalization and irreversible downregulation of IgLON5. Researchers therefore addressed the question of whether the mechanism of action of the antibodies by downregulating IgLON5 disrupted the interaction in the internal cytoskeleton, thereby inducing tau accumulation. This hypothesis was confirmed by a Barcelona research group last year, the expert said. Neurodegenerative changes with ring structures, early terminations of dendrites as well as bundles could be detected. In addition, the induction of a dew accumulation could be proven. These effects were observed after three weeks, which in conclusion means that IgLON5 antibody-associated tauopathy is a slowly and chronically progressive disease. It represents an important differential diagnosis to primary neurodegenerative diseases. The phenotypic spectrum of IgLON5 tauopathy is wide (Table 1) .
Another important aspect is CASPR2 antibodies. For example, they may be associated with late-onset ataxia in adulthood. Especially when this is associated with pain and epileptic seizures. In addition, a characteristic phenotype is the presence of CASPR2 antibodies in leg myoclonus. This primarily affects men in middle or later life with neuropathic pain, fasciculations, cognitive impairment, or epileptic seizures.
The speaker summarized that in adulthood especially the antibodies LGI1, IgLON5 and CASPR2 play a major role. The most characteristic phenotypes include FBDS, NREM parasomnia and leg myoclonus. Accordingly, hyponatremia should be thought of as LGI1, sleep-associated movement disorders and swallowing/breathing/oculomotor dysfunction as IgLON5, and neuropathic pain and myokymia as CASPR2.
Source: “Rare but treatable movement disorders”, 07.05.2022, Prof. Dr. med. Bettina Ballint, Zurich, FomF Neurology Update Refresher, 06.-07.05.2022, Zurich and online.
InFo NEUROLOGY & PSYCHIATRY 2022; 20(3): 38.









