T cells that are endowed with tumor specificity through the expression of a chimeric antigen recept or (CAR) are becoming increasingly important. They are already increasingly being used in adoptive cell therapy in the fight against cancer. The major advantage of transferring a CAR as opposed to transferring a normal T-cell recept or (TCR) is that a CAR can recognize the tumor independently of the MHC.
T cells that are endowed with tumor specificity through the expression of a chimeric antigen recept or (CAR) are becoming increasingly important. They are already increasingly being used in adoptive cell therapy in the fight against cancer. The CAR concept was originally developed in the late 1980s by Zelig Eshhar (Weizmann Institute of Science, Rehovot, Israel) [1,2]. Most CARs consist of a tumor antigen-binding, antibody-derived scFv construct (single chain variable fragment, which is an artificially produced fusion protein consisting of a variable part of a light and a heavy chain of an immunoglobulin) and the intracellular part of the CD3ζ chain, the in c sharp is linked to one or more costimulatory domains [3]. This building block-like structure enables antigen-specific T cell activation in response to the specific recognition of antigens on the surface of malignant cells, initiated by the binding of scFv, and subsequent signaling via the CD3ζ chain as well as via the costimulatory domain [3]. Costimulation usually occurs either via CD28 (immunoglobulin superfamily) or via 4-1BB (TNF receptor superfamily) [3]. However, there are also many other formats. The major advantage of transferring a CAR as opposed to transferring a normal T-cell recept or (TCR) is that a CAR can recognize the tumor independently of the MHC.
This technology has so far been used to develop CARs that target various cell surface antigens on solid or hematological tumors. CAR-T cells, specific for target antigens such as CD19 on B cells or B cell maturation antigen (BCMA) on plasma cells, led to impressive clinical regressions in leukemias, lymphomas or myelomas in several clinical studies [4–6]. Results such as these led, among other things the approval of tisagenlecleucel for the treatment of acute B-cell lymphoblastic leukemia (ALL), axicabtagene-ciloleucel for the treatment of aggressive B-cell non-Hodgkin’s lymphoma, Brexucabtagene-Autoleucel for the treatment of mantle cell lymphoma, Lisocabtagene-Maraleucel for the treatment of large B-cell lymphoma and Idecabtagene-Vicleucel and Ciltacabtagene-Autoleucel for the treatment of multiple myeloma by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) [3].
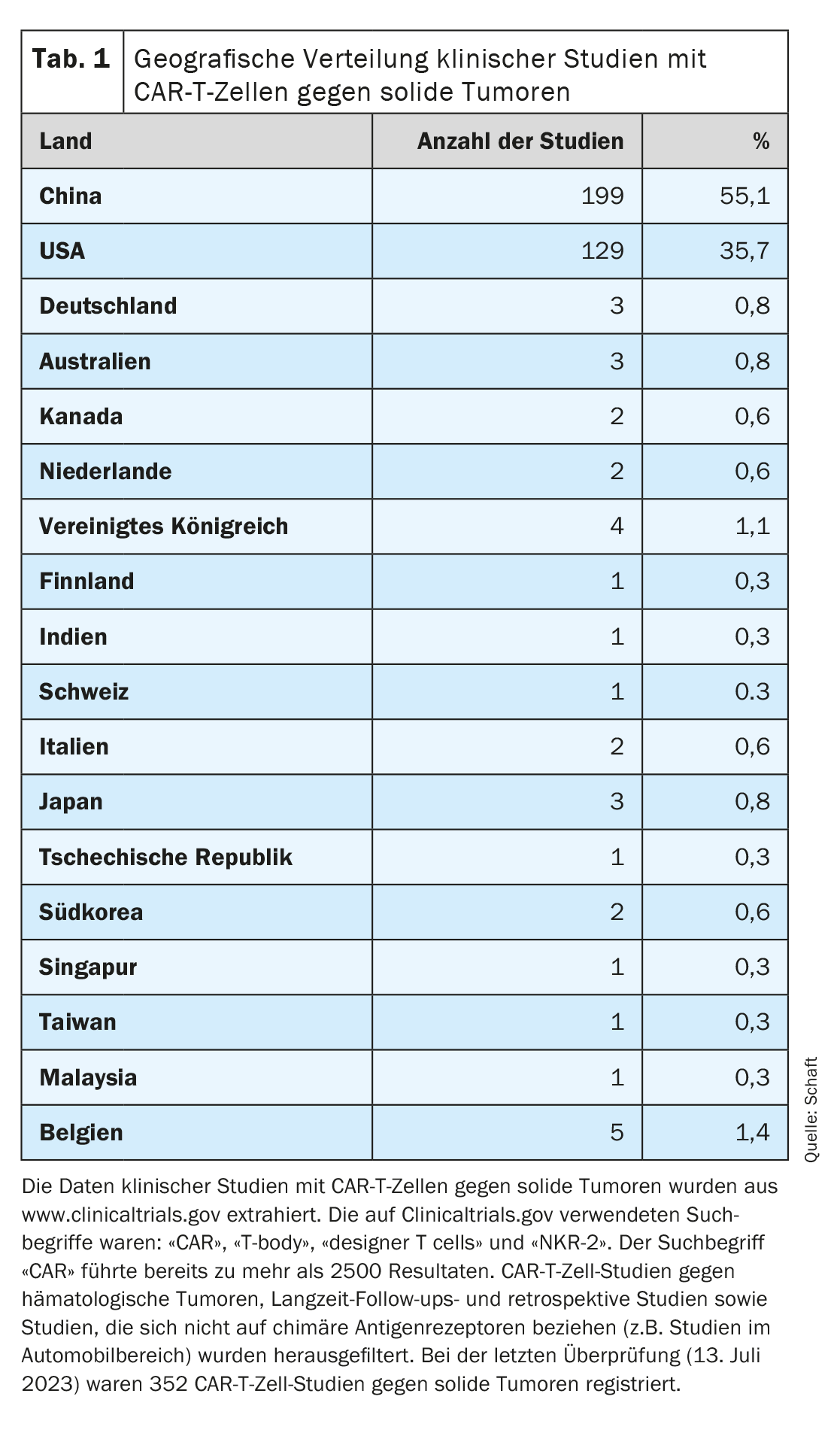
Since most clinical trials focus on the elimination of hematological tumors, the development of CAR-T cells against solid tumors is lagging behind (detailed overview in [7–11]). Looking at the geographical distribution of clinical trials with CAR-T cells against solid tumors registered on Clinicaltrials.gov (n=352; last assessment on July 13, 2023), it is clear that most of these trials are conducted in China (n=199; 55.1%). The USA is in second place (n=129; 35.7%). Only very few studies are conducted in Europe (Germany n=3, Switzerland n=1), Australia and the rest of Asia (combined n=33; 9.2%) (Table 1).
CAR formats
Since the publication of the first CAR concept by Zelig Eshhar in 1989 [1,2], CARs have been continuously developed. This resulted in several generations of CARs based on the basic framework of the original CAR concept. The classic CAR always contains an antibody-based scFv that can bind the tumor antigen. In first-generation CARs (Fig. 1), the scFv is connected to either the intracellular signaling domain of FcεRIγ or CD3ζ via a flexible linker and a transmembrane domain [11,12].
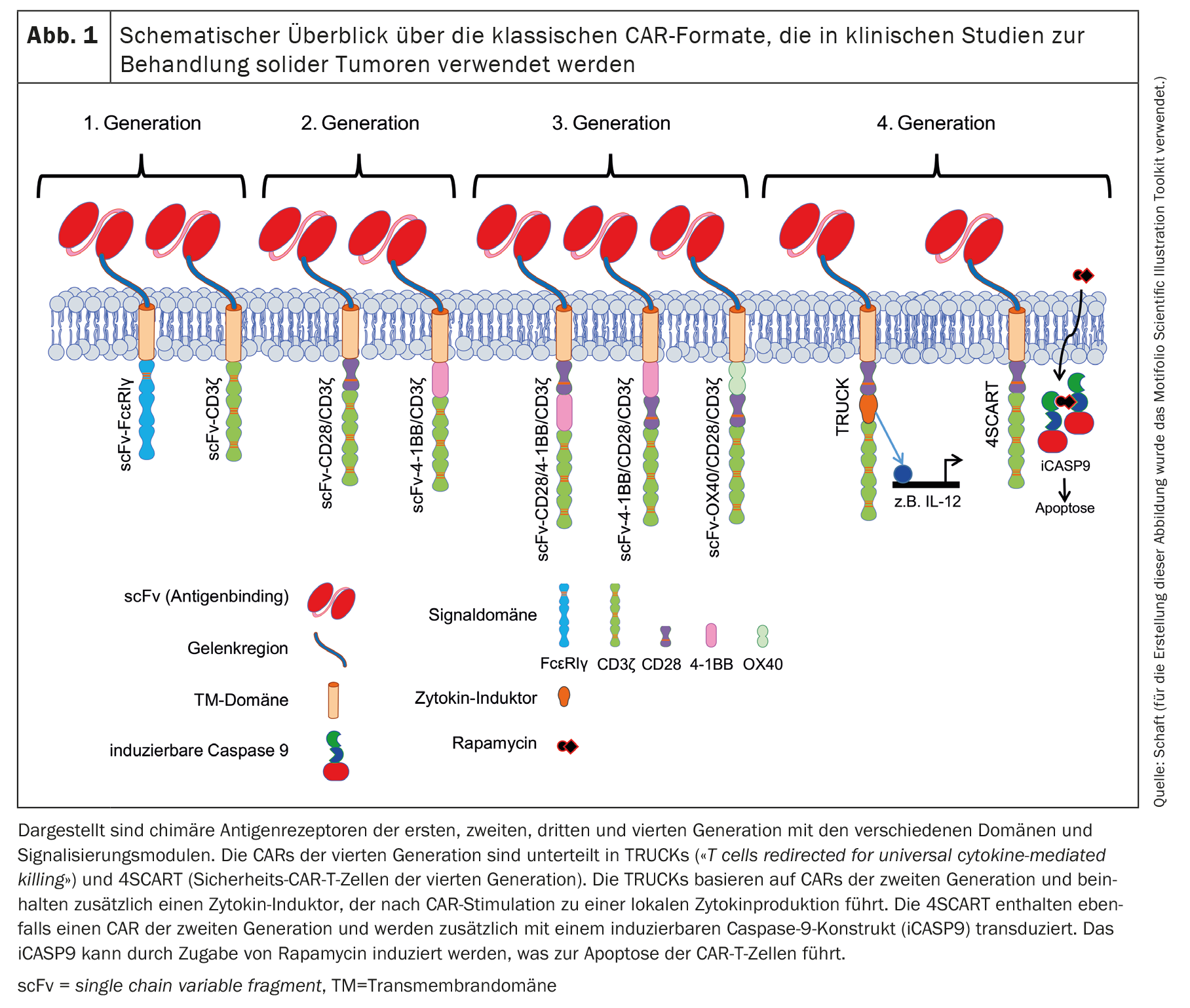
Most registered clinical trials with CAR-T cells against solid tumors use a second-generation CAR [11,12], which also contains a costimulatory domain (Fig. 1). The costimulation is usually performed by CD28 or 4-1BB [3]. CD28 co-stimulation physiologically supports the production of IL-2, -6, -10 and other interleukins as well as cell cycle progression, survival, differentiation and cytolytic function of T cells [13]. In many studies in which CARs with a CD28 signaling domain were used, effective and rapid antitumor effects were observed. However, these were only of short duration and were associated with limited survival in vivo compared to CARs with a 4-1BB signaling domain, for example [14]. Physiological 4-1BB signaling in T cells enhances cell cycle progression and proliferation, cytokine secretion, cytolytic potential of T cells and inhibits clonal deletion andactivation-induced cell death (AICD) [15,16]. CARs containing 4-1BB as a signaling domain not only enabled more robust cell activation, increased persistence in vivo, but also promoted the differentiation of CAR-T cells towardscentral memory cells [4,14,17–24].
The third-generation CARs [11,12] contain combinations of costimulatory domains: CD28/4-1BB, 4-1BB/CD28 or OX40/CD28 (Fig. 1) [25,26]. Fourth-generation CARs are basically second-generation CARs with additional features. TRUCKs (T cells redirected for universal cytokine-mediated killing) are modified to produce cytokines in a very limited localized manner [27]. The induced effects depend on the type of cytokines released: IL-12, for example, can activate an innate immune response against the tumor [28], causes reduced susceptibility to inhibitory effects by regulatory T cells (Tregs) [29] and increases cytokine secretion and T cell proliferation [30,31]. IL-15, on the other hand, increases the antitumor activity of CAR-T cells [32].
Another variant of the fourth generation is the 4SCART (safety CAR-T cells). These T cells are transduced simultaneously with a CAR and an inducible caspase 9 (iCASP9) as a safety precaution against adverse events. iCASP9 can be induced by the addition of rapamycin, which leads to apoptosis of the CAR-T cells.
Transfer technologies
An essential necessity in the production of CAR T cells is to find a suitable method for transferring the CAR into the T cells. Various existing methods can be used for this purpose. Most clinical trials use a viral transfer method (retroviral or lentiviral) to stably introduce the CAR into the T cells. During this process, a CAR-encoding gene is transported from the virus into the T cell, where it is stably integrated into the genomic DNA. The progeny of these transduced cells all carry the CAR gene and can express the receptor on their cell surface. Disadvantages of viral transduction are the random integration into the genome of the host cell, which can lead to the destruction or activation of some genes (i.e. insertional mutagenesis), as well as the introduction of viral material/genes. This method can lead to serious side effects in patients treated with CAR-T cells. Lamers et al. described, for example, the development of immune responses to the receptor-encoding transgene and the retroviral vector [33].
Some clinical trials use a non-viral gene delivery system or a transfer method that integrates the CAR gene at a specific site (e.g. Sleeping Beauty transposon system [34–37], PiggyBac transposon system [36,37], CRISPR-Cas9 [38]). The transfection of DNA or RNA are further transfer systems [39], which, however, do not lead to the integration of the CAR-coding sequence into the genome of the host cell. The resulting transient expression of the CAR has certain advantages.
CAR-T cells against solid tumors – antigen selection and safety precautions
As described above, the clinical use of CAR-T cells in the treatment of solid tumors lags behind the success of CAR-T cells in the treatment of hematological tumors. One of the reasons for this is that CD19 and BCMA are target antigens that are specifically expressed by B cells or plasma cells and their complete elimination is relatively harmless. Other antigens, especially on solid tumors, are often also expressed on healthy tissue, which makes it difficult to select a suitable target antigen.
Selection of the antigen
Ideal target antigens on solid tumors combine three essential properties:
- Uniform expression on the surface of malignant cells, which reduces the risk of antigen-negative escape variants.
- A lack of expression on non-malignant cells (i.e. exclusive expression on the tumor cells), thereby preventing the risk of serious, potentially fatal side effects arising from on-target/off-tumor activity of the CAR-T cells [40,41].
- A crucial role as an oncogenic driver in cancer cells that prevents antigen downregulation due to a selective survival advantage of malignant cells.
- In addition, the co-expression of the antigen on neighboring cells within the tumor microenvironment (e.g. on tumor-associated vessels, fibroblasts and macrophages) is another positive characteristic of an ideal target antigen, as the supply structure of the tumor can be attacked by the antigen-specific therapy.
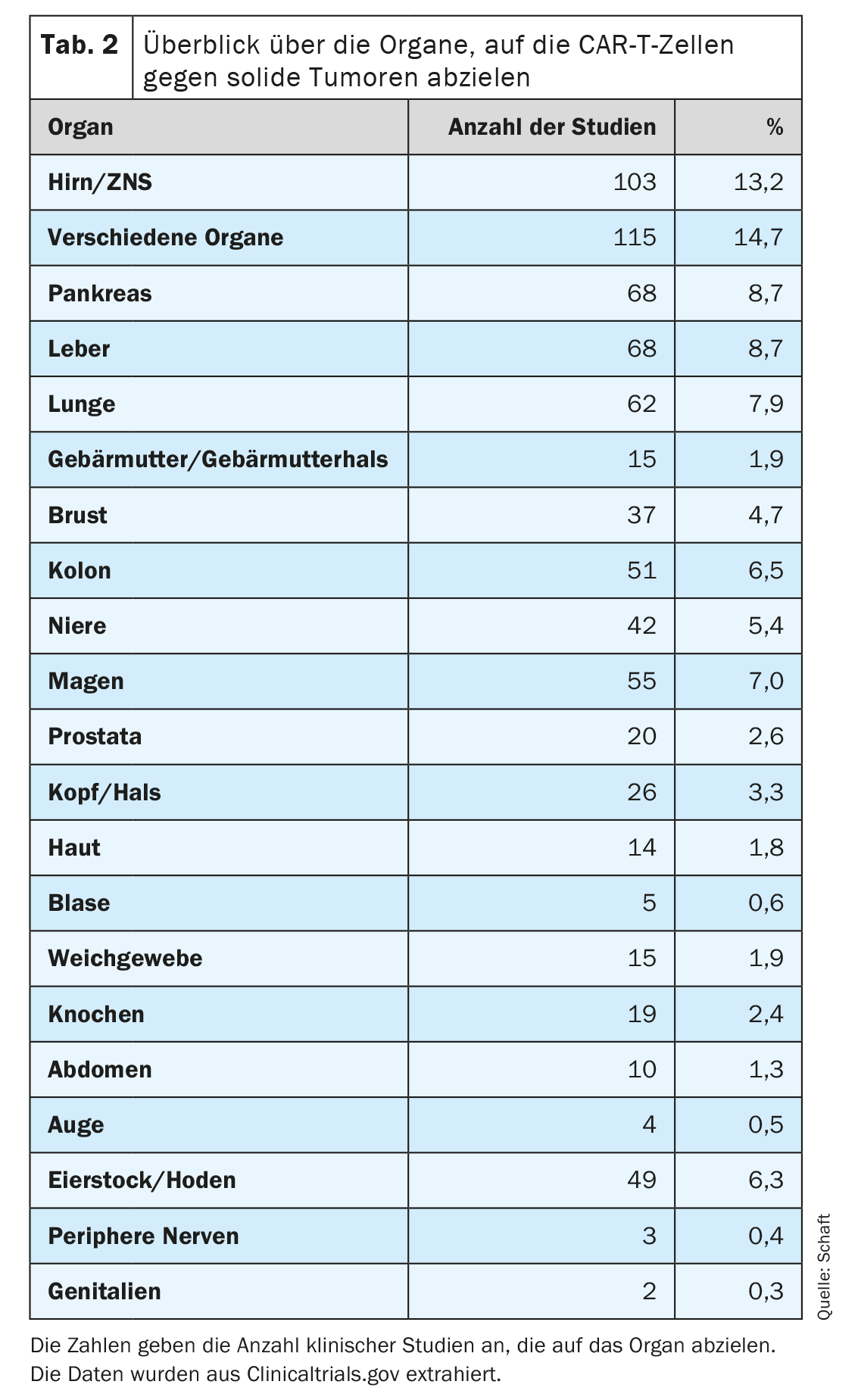
The second point in particular poses the greatest problem for the development of CAR-T cells against solid tumors, since most antigens expressed on solid tumors are also expressed on important healthy tissue. This can lead to an undesirable on-target/off-tumor reaction and associated side effects. Nevertheless, many different solid tumor types (81 cancer entities in total) in a total of 20 organs are being targeted with CAR-T cells specific for 63 different antigens (Table 2). In many clinical studies, tumors of the brain/CNS, liver, pancreas and lung in particular are being investigated (n=103, 68, 68 and 62, respectively; Tab. 2). This could be due to the high medical need and/or the lack of effective alternative therapies for tumors in the corresponding organs.
Safety precautions
If a target antigen is recognized by transferred CAR-T cells on healthy tissue, undesirable, severe side effects can occur. Several strategies have been developed to be able to switch off the CAR-T cells as quickly as possible in the event of toxicity in the patient. Rapamycin, a molecule capable of inducing dimerization of constructs, can be used, for example, to activate an inducible caspase 9. In 4SCART, these inducible constructs are transferred into the T cells simultaneously with the CAR as a so-called suicide switch (Fig. 1). After rapamycin-induced dimerization, caspase 9 induces apoptosis of the CAR T cells. This also eliminates the unwanted/unexpected T cell activity against healthy tissue(on-target/off-tumor effects) [42,43]. Other possible switches, such as the herpes simplex virus thymidine kinase/ganciclovir (HSV-tk/GCV) strategy [44,45] are already being used [11].
A special safety measure to circumvent prolonged autoimmunity induced by an on-target/off-tumor reaction of the CAR is the transfection of the CAR by mRNA electroporation [11]. We have already shown in several publications that transient transfection of T cells with CARs using mRNA electroporation can be an effective and safe tool in cancer immunotherapy [46-50]. The electroporation process is based on complex physico-chemical mechanisms that lead to perforation of the plasma membrane through the application of electric fields and enable the subsequent entry of mRNA into the cytosol [51]. The use of RNA-transfected CAR-T cells offers the advantage that receptor expression is limited in time, so that potential off-target and on-target/off-tumor toxicity is also limited. The CAR-RNA transfer strategy is particularly attractive in phase 0/1 clinical trials investigating novel tumor antigens for CAR T-cell therapy with an unknown clinical safety profile.
Clinically tested CAR-T cells against uveal melanoma
Surprisingly, only four clinical CAR T-cell studies against solid tumors focus on the eye entity (Table 2) . Of these, two studies are directed against retinoblastoma and two studies against uveal melanoma. Uveal melanoma is the most common type of eye cancer and metastasizes in up to 50% of patients. Metastases occur predominantly in the liver and are associated with a poor median survival time of around 12 months. Despite tremendous progress in the treatment of metastatic cutaneous melanoma with immune checkpoint blockade (ICB), it shows no efficacy in uveal melanoma. Only the recently approved bispecific T-cell engager Tebentafusp (a CD3-specific scFv linked to a soluble TCR that recognizes a gp100 peptide presented by HLA-A2) can attenuate progression and prolong overall survival in a subset of patients with metastatic uveal melanoma. The observed positive effects of Tebentafusp are short-lived, with a median overall survival time of 22 months and a three-year survival rate of 24%. In addition, only 50% of metastatic patients are eligible for this treatment option due to HLA-A2 restriction. Therefore, there is also a high medical need for alternative treatment approaches for this tumor entity.
Currently, two clinical trials with CAR-T cells listed in the international US National Library of Medicine (www.clinicaltrials.gov) are recruiting patients with uveal melanoma (NCT03893019 against Cluster of Differentiation 20 [CD20] and NCT03635632 against disialoganglioside GD2 [GD2]). Both studies use CAR-T cells that target non-melanoma-specific antigens.
The first clinical trial (Phase 1; NCT03893019), which uses second-generation CD20-specific CAR-T cells (Fig. 2A), is sponsored by Miltenyi Biomedicine GmbH (Principal Investigator [PI]: Peter Borchmann; University Hospital Cologne) and is mainly recruiting patients with cutaneous melanoma. Some patients with uveal melanoma are also treated. CD20 is a target antigen that is expressed on normal B cells and is typically used as a target antigen in B-cell non-Hodgkin’s lymphoma [52]. However, it is also expressed on a small subset of melanoma cells [53,54]. However, targeting an antigen that is only expressed on a small subset of cancer cells could result in the tumor easily escaping CAR-T cell therapy. Unfortunately, the current status of this clinical trial is not known.
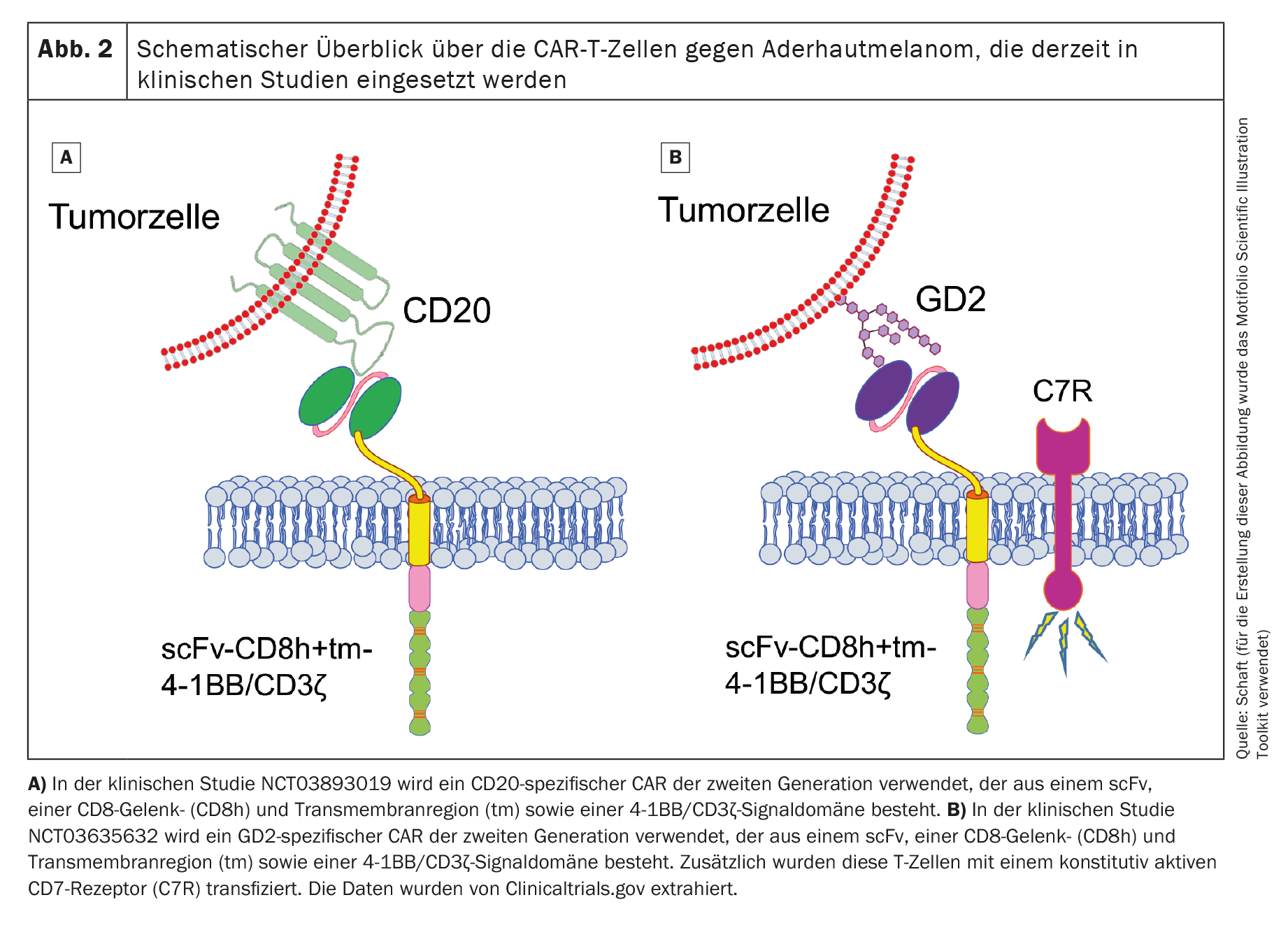
The second clinical trial (also phase 1; NCT03635632) uses GD2-specific CAR-T cells (Fig. 2B) and recruits patients with neuroblastoma, sarcoma, uveal melanoma, breast cancer or other cancers that express GD2. This study, sponsored by Baylor College of Medicine (PI: Bilal Omer; Baylor College of Medicine), is currently active, but no patients are currently being recruited.
In addition to the second-generation CAR, the researchers also transduce a constitutively active IL-7 receptor into the T cells in order to prolong the survival of the CAR-T cells after adoptive transfer. GD2 is expressed, albeit at very low levels, in the cerebellum and peripheral nerves [55], which makes treatment with GD2-specific CAR-T cells very risky if an on-target/off-tumor reaction is induced. No data has yet been published for this clinical trial either.
In summary, there is great potential for clinical trials with CAR-T cells against (uvea) melanoma-specific antigens where the risk of an on-target/off-tumor reaction is low.
The search for a better tumor antigen in uveal melanoma
The prevention or reduction of a possible on-target/off-tumor reaction is, as already mentioned in the section “CAR-T cells against solid tumors”, a basic requirement in the search for new antigens. Preclinically, the focus is currently on two antigens expressed on uveal melanomas: human epidermal growth factor receptor 2 (HER2) and chondroitin sulphate proteoglycan 4 (CSPG4).
HER2
HER2 is a member of the ErbB family of receptor tyrosine kinases (EGFR [ErbB-1], HER2 [/neu] [ErbB-2], Her 3 [ErbB-3] and Her 4 [ErbB-4]). Mutations in HER2 lead to overexpression, which leads to constitutive activation and the resulting uncontrolled cell division. This applies above all to breast cancer, but also to other types of cancer such as ovarian cancer or gliomas [56–58].
As already mentioned, the use of CAR-T cells is a double-edged sword, as the effectiveness of these cells can also turn against the patient [59]. It can never be ruled out that a rare but essential cell type expresses the antigen in healthy tissue. Researchers at the National Cancer Institute reported a case that illustrates the deadly potential of the on-target/off-tumor toxicity of the HER2 antigen. Shortly after infusion of HER2-specific CAR-T cells, clinical symptoms of acute respiratory distress syndrome requiring mechanical ventilation were observed in a patient with metastatic colorectal cancer [60]. Unfortunately, the patient died five days after the onset of acute respiratory distress [60]. The cause of death was probably the result of on-target/off-tumor toxicity caused by low levels of HER2 on epithelial cells in the lungs. Remarkably, the CAR was based on the FDA-approved monoclonal antibody trastuzumab, which has been used extensively without causing severe pulmonary toxicity [61]. This underlines the need for very careful selection of the target antigen for CAR T-cell therapy.
CSPG4
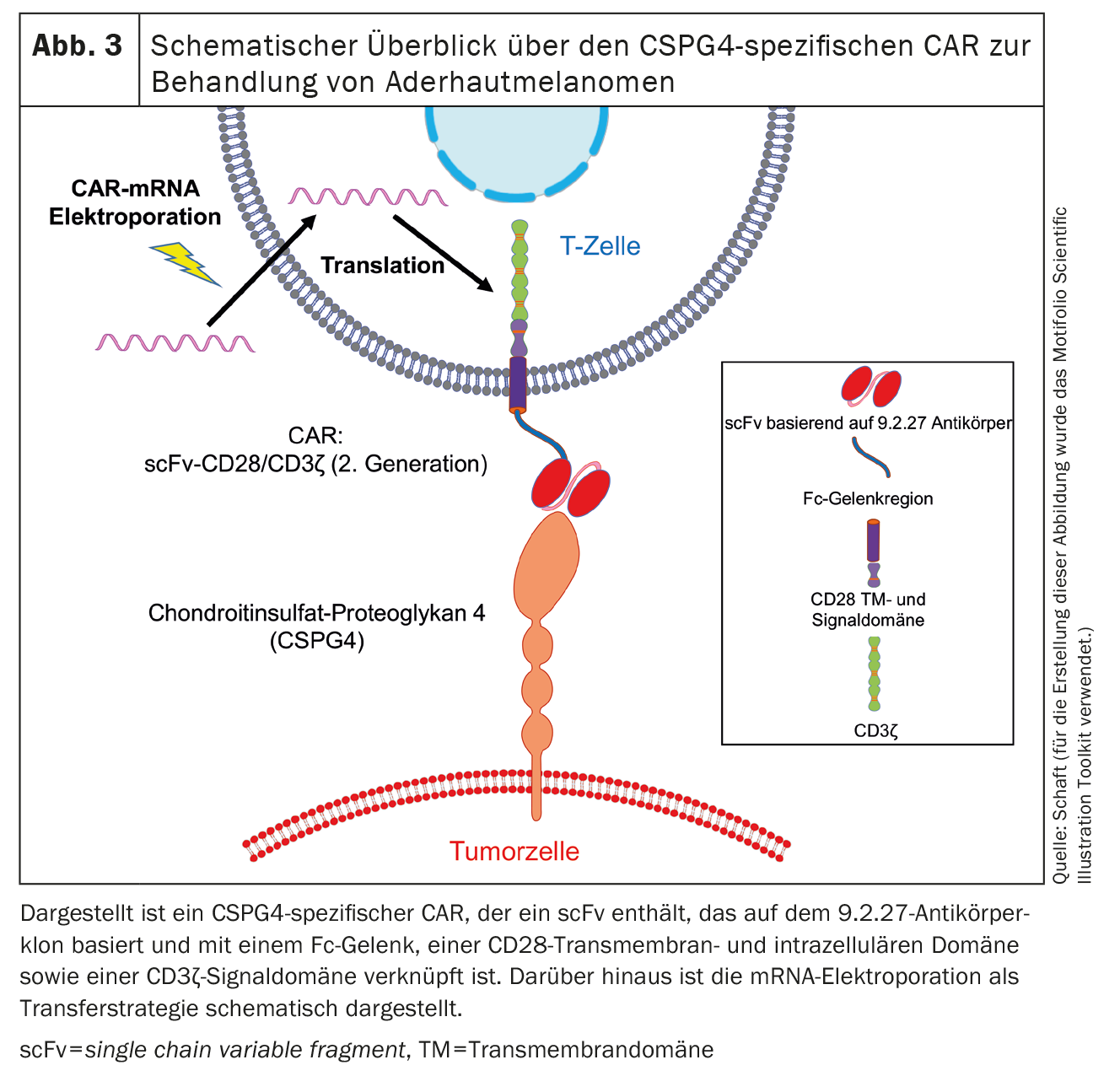
The second antigen expressed on uveal melanomas is chondroitin sulphate proteoglycan 4 (CSPG4) (Fig. 3), formerly also known as melanoma-associated chondroitin sulphate proteoglycan (MCSP) or high molecular weight melanoma-associated antigen (HMW-MAA). CSPG4 is a type 1 single-pass transmembrane protein and was discovered by Ralph Reisfeld [72]. We and other working groups [47-50, 62-71] worked mainly on CSPG4. CSPG4 expression is associated with increased proliferation and survival of tumor cells. This is initiated by the activation of the MAPK signaling pathway and the cross-presentation of growth factors [73]. In addition, CSPG4 plays a role in cell motility and tissue infiltration due to its association with the actin cytoskeleton and binding to various integrins and components of the extracellular matrix [74]. Furthermore, CSPG4 is involved in placenta formation [75], angiogenesis [76], the formation of neuronal networks [77], keratinocyte turnover and the homeostasis of epidermal stem cells [78].
Several publications described the expression of CSPG4 on non-pathological tissues, such as precursors of hair follicle and epidermal cells, endothelial cells and activated pericytes (but not on mature vessels) [79,80], chondrocytes of articular cartilage [81], smooth muscle cells [82] and cells of the neuromuscular synapse of human postnatal skeletal muscle [83]. However, CSPG4 is expressed significantly less on healthy tissue than on tumor cells [62,73,84].
Beard et al. showed that CSPG4 could be detected at the RNA level in a variety of normal tissues, including the central nervous system, eye, skin, adipose tissue, blood vessels, bladder, gastrointestinal tract, uterus, prostate, spleen and thymus [85]. On average, CSPG4 RNA is overexpressed 6.6-fold in malignant tissue (melanoma) compared to healthy tissue [85]. These results confirmed earlier work by Erfurt et al. which showed that while CSPG4 mRNA could be detected in some normal tissue samples, expression was significantly increased in cutaneous melanoma and uveal melanoma samples [86].
Immunohistochemical staining and reversed-phase protein arrays showed that specific CSPG4 expression at the protein level could only be detected in some samples from the small intestine [63]. No CSPG4 protein expression could be detected in the following tissues: Brain, peripheral nerves, skin, mesothelium, breast, heart, kidney, adrenal glands, liver, lung, lymph nodes, muscles, ovaries, pancreas, esophagus, prostate, spleen, stomach, uterus and thyroid [62,63]. In contrast, CSPG4 is expressed on almost all cutaneous melanoma cells [87–90]. Choroidal melanomas [91,92] and some other tumors such as sarcomas, astrocytomas, gliomas, neuroblastomas [93-96], leukemias [97–101] and triple-negative breast cancer also express CSPG4 [102]. In many of these malignancies, CSPG4 expression is associated with a poor prognosis and aggressive tumor growth [103].
In addition, CSPG4 is considered a primary tumor target antigen [84], as it plays a role in melanoma metastasis [104] and is expressed on activated pericytes during angiogenesis in tumors as well as in hypoxia [105–107]. The latter enables targeting of the tumor vasculature. Most importantly, CSPG4 acts as an oncogenic driver in melanoma and promotes the growth and survival of malignant cells after activation of different signaling pathways [73]. Therefore, the tumor cannot simply downregulate CSPG4 to escape CSPG4-targeted therapy.
For this reason, CSPG4 has already been selected as a target antigen by several groups and CSPG4-specific CARs have been introduced into the T cells via various mechanisms. CSPG4-specific CARs of different formats that were virally transduced into T cells resulted in strong cytotoxicity of T cells in vitro . In animal models, the adoptively transferred T cells reacted to various CSPG4-expressing tumors, such as melanoma, breast cancer, mesothelioma, glioblastoma and osteosarcoma [47–50,62–71]. Geldres et al. retrovirally transduced a second-generation CSPG4-specific CAR into T cells. In vitro, these CSPG4-specific CAR T cells were able to recognize and lyse melanoma cells in an antigen-specific manner [65]. In addition, binding of the antigen to the CAR led to a pronounced secretion of IL-2 and IFNγ. In vivo , the transfer of CSPG4-specific CAR-T cells into melanoma cell-bearing mice led to a significant slowdown in tumor growth and an improvement in the overall survival of the mice [65]. In the same publication, the positive lack of reactivity of CSPG4-specific CAR-T cells to normal tissue with detectable CSPG4 RNA but without CSPG4 protein expression was described [65]. They were able to show that CSPG4-specific CAR-T cells did not exert significant cytotoxicity against primary epithelial cell lines from the prostate, lung and kidney [65]. Therefore, the expression analyses by Beard et al. [63,85], mitigate the concerns already described above regarding CSPG4-specific CAR-T cell-inducedon-target/off-tumor toxicity, as expression of CSPG4 at the protein level is required to induce unwanted CAR-T cell reactivity.
So far, we have used the method of mRNA transfection by electroporation to introduce CARs into T cells. In the course of this, we have already tested several CSPG4-specific CARs and observed that mRNA-transfected CAR-T cells are able to eliminate tumor cells in an antigen-specific manner. The expression kinetics of CARs transferred by electroporation depends on the CAR backbone [47]. A CAR was identified that exhibits both high expression on T cells and high anti-tumor cell reactivity. This CAR contains a scFv based on the 9.2.27 antibody clone linked to an Fc spacer, a CD28 transmembrane and intracellular domain and a CD3ζ signaling domain [47](Fig. 3). In vivo experiments with immunodeficient Rag-/-/ common γ-chain-/- mice showed that transfected CSPG4-specific CAR-T cells significantly prolonged the mean survival time of the mice [47].
In order to transfer the CSPG4-specific CAR-T cells into clinical application, the production of CAR-T cells on a clinical scale was established in the laboratory by mRNA transfection of a CAR in full GMP compliance [50]. This showed that the repeated production of a sufficient number of highly pure CSPG4-specific CAR-transfected T cells is possible. These modified T cells show very high transfection efficiency, high CAR expression and high efficacy in killing melanoma target cells [50].
Although CSPG4 is a primary tumor target antigen, especially in cutaneous melanomas, we also found that it was not expressed on several uveal melanoma cell lines we tested. We have therefore established a combined in silico/in vitro platformto identify new tumor-specific cell surface antigens of uveal melanomas. Using this platform, we have identified a candidate protein as a suitable target antigen in uveal melanoma for the development of further CARs. The generated CARs that can bind to this candidate protein are currently being tested for their functionality and specificity.
Outlook
The use of CAR-T cells in the treatment of solid tumors offers great opportunities. Further preclinical studies and clinical trials are required to respond to the high medical need for treatment of solid cancer entities (such as uveal melanoma).
Future clinical trials should focus on testing new CAR formats. In addition to testing new extracellular antigen-binding domains and new intracellular signaling domains [109], this also includes testing formats that increase the safety of CAR-T cell use [108]. In addition, new cellular vehicles for CAR transfer [109,110] promise to expand the range of applications. For example, the possibility of standard use of CAR-NK cells [111] or allogeneic CAR-T cells [112] can reduce the costs of CAR cell therapy and thus make the therapy accessible to more patients.
In addition, more tumor-specific antigens should be found in order to prevent on-target/off-tumor reactions. Antigens that are expressed on the tumor stroma and can be attacked there by CAR-T cells are very promising in this area [113]. The targeting of multiple antigens by a CAR-T cell (i.e. the expression of different CARs specific for different antigens on a single cell) can increase tumor specificity and reduce the risk of off-target effects. This also applies to the model when intracellular signaling modules are shared between the different CARs to increase the safety profile of the CAR-T cells. This also makes the development of antigen loss variants of the tumors less likely.
Furthermore, combination therapies of CAR-T cells with various so-called small molecules or monoclonal antibodies to prevent tumor escape mechanisms and to increase antitumor activity are already being clinically tested in many hematological tumors (detailed overview in [114,115]). Such combinations are also promising for the treatment of solid tumors and must be tested in clinical trials in the near future.
N.S. conducted the research on clinicaltrials.gov, revised the figures and drafted the manuscript. S.H. edited the manuscript. N.S. and S.H. jointly drafted the CME training questions.
Literature:
- Gross G, Gorochov G, Waks T, Eshhar Z: Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc 1989; 21(1 Pt 1): 127-130.
- Gross G, Waks T, Eshhar Z: Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989; 86(24): 10024-10028.
- June CH, Sadelain M: Chimeric Antigen Receptor Therapy. N Engl J Med 2018; 379(1): 64-73.
- Maude SL, Laetsch TW, Buechner J, et al: Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018; 378(5): 439-448.
- Neelapu SS, Locke FL, Go WY: CAR T-Cell Therapy in Large B-Cell Lymphoma. N Engl J Med 2018; 378(11): 1065.
- Schuster SJ, Bishop MR, Tam CS, et al: Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 2019; 380(1): 45-56.
- Wang Z, Guo Y, Han W: Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 2017; 8(12): 896-925.
- Han S, Latchoumanin O, Wu G, et al: Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett 2017; 390: 188-200.
- Yeku O, Li X, Brentjens RJ: Adoptive T-Cell Therapy for Solid Tumors. Am Soc Clin Oncol Educ Book 2017; 37: 193-204.
- Arabi F, Torabi-Rahvar M, Shariati A, et al: Antigenic targets of CAR T Cell Therapy. A retrospective view on clinical trials. Exp Cell Res 2018; 369(1): 1-10.
- Schaft N: The Landscape of CAR-T Cell Clinical Trials against Solid Tumors-A Comprehensive Overview. Cancers (Basel) 2020; 12(9).
- Holzinger A, Abken H: CAR T Cells: A Snapshot on the Growing Options to Design a CAR. Hemasphere 2019; 3(1): e172.
- Boomer JS, Green JM: An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol 2010; 2(8): a002436.
- Kawalekar OU, O’Connor RS, Fraietta JA, et al.: Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016; 44(2): 380–390.
- Cannons JL, Choi Y, Watts TH: Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol 2000; 165(11): 6193–6204.
- Lee HW, Nam KO, Park SJ, Kwon BS: 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur J Immunol 2003; 33(8): 2133-2141.
- Zhao Z, Condomines M, van der Stegen SJC, et al: Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015; 28(4): 415-428.
- Milone MC, Fish JD, Carpenito C, et al: Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009; 17(8): 1453-1464.
- Lim WA, June CH: The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017; 168(4): 724-740.
- Roselli E, Frieling JS, Thorner K, et al: CAR-T Engineering: Optimizing Signal Transduction and Effector Mechanisms. BioDrugs 2019; 33(6): 647-659.
- Hombach AA, Holzinger A, Abken H.: The weal and woe of costimulation in the adoptive therapy of cancer with chimeric antigen receptor (CAR)-redirected T cells. Curr Mol Med 2013; 13(7): 1079-1088.
- Sadelain M, Brentjens R, Riviere I: The basic principles of chimeric antigen receptor design. Cancer Discov 2013; 3(4): 388-398.
- Redeker A, Arens R: Improving Adoptive T Cell Therapy: The Particular Role of T Cell Costimulation, Cytokines, and Post-Transfer Vaccination. Front Immunol 2016; 7: 345.
- Weinkove R, George P, Dasyam N, McLellan AD: Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology 2019; 8(5): e1049.
- Ceppi F, Rivers J, Annesley C, et al: Lymphocyte apheresis for chimeric antigen receptor T-cell manufacturing in children and young adults with leukemia and neuroblastoma. Transfusion 2018; 58(6): 1414-1420.
- Li W, Guo L, Rathi P, et al: Redirecting T Cells to Glypican-3 with 4-1BB Zeta Chimeric Antigen Receptors Results in Th1 Polarization and Potent Antitumor Activity. Hum Gene Ther 2017; 28(5): 437-448.
- Chmielewski M, Hombach AA, Abken H: Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 2014; 257(1): 83-90.
- Chmielewski M, Kopecky C, Hombach AA, Abken H.: IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively pattern an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res 2011; 71(17): 5697-5706.
- Pegram HJ, Lee JC, Hayman EG, et al: Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012; 119(18): 4133-4141.
- Koneru M, Purdon TJ, Spriggs D, et al: IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015; 4(3): e994446.
- Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ: A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med 2015; 13: 102.
- Xu A, Bhanumathy KK, Wu J, et al: IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci 2016; 6: 30.
- Lamers CH, Willemsen R, van Elzakker P, et al.: Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011; 117(1): 72–82.
- Magnani CF, Tettamanti S, Alberti G, et al.: Transposon-Based CAR T Cells in Acute Leukemias: Where are We Going? Cells 2020; 9(6).
- Hudecek M, Ivics Z: Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr Opin Genet Dev 2018; 52: 100-108.
- Tipanee J, VandenDriessche T, Chuah MK: Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum Gene Ther 2017; 28(11): 1087-1104.
- Vargas JE, Chicaybam L, Stein RT, et al: Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J Transl Med 2016; 14(1): 288.
- Ran FA, Hsu PD, Wright J, et al: Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8(11): 2281-2308.
- Birkholz K, Hombach A, Krug C, et al: Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther 2009; 16(5): 596-604.
- Lamers CH, Sleijfer S, et al: Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 2013; 21(4): 904-912.
- Morgan RA, Yang JC, Kitano M, et al: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18(4): 843-851.
- Stavrou M, Philip B, Traynor-White C, et al: A Rapamycin-Activated Caspase 9-Based Suicide Gene. Mol Ther 2018; 26(5): 1266-1276.
- Di Stasi A, Tey SK, Dotti G, et al: Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365(18): 1673-1683.
- Moolten FL.: Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res 1986; 46(10): 5276-5281.
- Beltinger C, Fulda S, Kammertoens T, et al: Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci U S A 1999; 96(15): 8699-8704.
- Harrer DC, Simon B, Fujii SI, et al: RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: a safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017; 17(1): 551.
- Krug C, Birkholz K, Paulus A, et al: Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol Immunother 2015; 64(12): 1623-1635.
- Dörrie J, Babalija L, Hoyer S, et al: BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int J Mol Sci 2018; 19(1).
- Harrer DC, Schuler G, Dörrie J, Schaft N: CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia. Int J Mol Sci 2019; 20(11).
- Wiesinger M, Marz J, Kummer M, et al: Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers (Basel) 2019; 11(8).
- Shi J, Ma Y, Zhu J, et al: A Review on Electroporation-Based Intracellular Delivery. Molecules 2018; 23(11).
- Ernst M, Oeser A, Besiroglu B, et al: Chimeric antigen receptor (CAR) T-cell therapy for people with relapsed or refractory diffuse large B-cell lymphoma. Cochrane Database Syst Rev 2021; 9(9): Cd013365.
- Pinc A, Somasundaram R, Wagner C, et al: Targeting CD20 in melanoma patients at high risk of disease recurrence. Mol Ther 2012; 20(5): 1056-1062.
- 54 Schmidt P, Kopecky C, Hombach A, et al: Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A 2011; 108(6): 2474-2479.
- Brignole C, Marimpietri D, Pagnan G, et al: Neuroblastoma targeting by c-myb-selective antisense oligonucleotides entrapped in anti-GD2 immunoliposome: immune cell-mediated anti-tumor activities. Cancer Lett 2005; 228(1-2): 181-186.
- Mitri Z, Constantine T, O’Regan R: The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother Res Pract 2012; 2012: 743193.
- Slamon DJ, Godolphin W, Jones LA, et al: Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244(4905): 707-712.
- Zhang JG, Kruse CA, Driggers L, et al: Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J Neurooncol 2008; 88(1): 65-76.
- Casucci M, Hawkins RE, Dotti G, Bondanza A: Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol Immunother 2015; 64(1): 123-130.
- Morgan RA, Yang JC, Kitano M, et al: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18(4): 843-851.
- Slamon DJ, Leyland-Jones B, Shak S, et al: Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344(11): 783-792.
- Wang Y, Geldres C, Ferrone S, Dotti G: Chondroitin sulfate proteoglycan 4 as a target for chimeric antigen receptor-based T-cell immunotherapy of solid tumors. Expert Opin Ther Targets 2015; 19(10): 1339-1350.
- Beard RE, Zheng Z, Lagisetty KH, et al: Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer 2014; 2: 25.
- Pellegatta S, Savoldo B, Di IN, et al: Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci Transl Med 2018; 10(430).
- Geldres C, Savoldo B, Hoyos V, et al: T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res 2014; 20(4): 962-971.
- Abken H, Hombach A, Heuser C, Reinhold U.: A novel strategy in the elimination of disseminated melanoma cells: chimeric receptors endow T cells with tumor specificity. Recent Results Cancer Res 2001; 158: 249-264.
- Burns WR, Zhao Y, Frankel TL, et al: A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res 2010; 70(8): 3027-3033.
- Losch FO, Muller R, Mutschler B, et al: Activation of T cells via tumor antigen specific chimeric receptors: the role of the intracellular signaling domain. Int J Cancer 2003; 103(3): 399-407.
- Reinhold U, Liu L, Ludtke-Handjery HC, et al: Specific lysis of melanoma cells by receptor grafted T cells is enhanced by anti-idiotypic monoclonal antibodies directed to the scFv domain of the receptor. J Invest Dermatol 1999; 112(5): 744-750.
- Schmidt P, Kopecky C, Hombach A, et al: Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A 2011; 108(6): 2474-2479.
- Harrer D, Simon B, Fujii SI, et al: RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: a safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017; 17: 551.
- Bumol TF, Reisfeld RA: Unique glycoprotein-proteoglycan complex defined by monoclonal antibody on human melanoma cells. Proc Natl Acad Sci USA 1982; 79(4): 1245-1249.
- Ilieva KM, Cheung A, Mele S, et al: Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front Immunol 2017; 8: 1911.
- Schiffer D, Mellai M, Boldorini R, et al: The Significance of Chondroitin Sulfate Proteoglycan 4 (CSPG4) in Human Gliomas. Int J Mol Sci 2018; 19(9).
- Van Sinderen M, Cuman C, Winship A, et al: The chrondroitin sulfate proteoglycan (CSPG4) regulates human trophoblast function. Placenta 2013; 34(10): 907-912.
- Fukushi J, Makagiansar IT, Stallcup WB: NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol Biol Cell 2004; 15(8): 3580-3590.
- Sakry D, Neitz A, Singh J, et al: Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol 2014; 12(11): e1001993.
- Legg J, Jensen UB, Broad S, et al: Role of melanoma chondroitin sulphate proteoglycan in patterning stem cells in human interfollicular epidermis. Development 2003; 130(24): 6049-6063.
- Ferrone S, Chen ZJ, Liu CC, et al: Human high molecular weight-melanoma associated antigen mimicry by mouse anti-idiotypic monoclonal antibodies MK2-23. Experimental studies and clinical trials in patients with malignant melanoma. Pharmacol Ther 1993; 57(2-3): 259-290.
- Schlingemann RO, Rietveld FJ, de Waal RM, et al: Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am J Pathol 1990; 136(6): 1393-1405.
- Midwood KS, Salter DM: Expression of NG2/human melanoma proteoglycan in human adult articular chondrocytes. Osteoarthritis Cartilage 1998; 6(5): 297-305.
- Tordsson JM, Ohlsson LG, Abrahmsen LB, et al: Phage-selected primate antibodies fused to superantigens for immunotherapy of malignant melanoma. Cancer Immunol Immunother 2000; 48(12): 691-702.
- Petrini S, Tessa A, Carrozzo R, et al: Human melanoma/NG2 chondroitin sulfate proteoglycan is expressed in the sarcolemma of postnatal human skeletal myofibers. Abnormal expression in merosin-negative and Duchenne muscular dystrophies. Mol Cell Neurosci 2003; 23(2): 219-231.
- Campoli MR, Chang CC, Kageshita T, et al: Human high molecular weight-melanoma-associated antigen (HMW-MAA): a melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit Rev Immunol 2004; 24(4): 267-296.
- Beard RE, Abate-Daga D, Rosati SF, et al: Gene expression profiling using nanostring digital RNA counting to identify potential target antigens for melanoma immunotherapy. Clin Cancer Res 2013; 19(18): 4941-4950.
- Erfurt C, Sun Z, Haendle I, et al: Tumor-reactive CD4+ T cell responses to the melanoma-associated chondroitin sulphate proteoglycan in melanoma patients and healthy individuals in the absence of autoimmunity. J Immunol 2007; 178(12): 7703-7709.
- Natali PG, Giacomini P, Russo C, et al: Antigenic profile of human melanoma cells. Analysis with monoclonal antibodies to histocompatibility antigens and to melanoma-associated antigens. J Cutan Pathol 1983; 10(4): 225-237.
- Berd D, Herlyn M, Koprowski H, Mastrangelo MJ: Flow cytometric determination of the frequency and heterogeneity of expression of human melanoma-associated antigens. Cancer Res 1989; 49(23): 6840-6844.
- Morgan AC Jr, Galloway DR, Reisfeld RA: Production and characterization of monoclonal antibody to a melanoma specific glycoprotein. Hybridoma 1981; 1(1): 27-36.
- Morgan AC Jr, Woodhouse C, Bartholemew R, Schroff R: Human melanoma-associated antigens: analysis of antigenic heterogeneity by molecular, serologic and flow-cytometric approaches. Mol Immunol 1986; 23(2): 193-200.
- Li Y, Madigan MC, Lai K, et al: Human uveal melanoma expresses NG2 immunoreactivity. Br J Ophthalmol 2003; 87(5): 629-632.
- Li Y, Wang J, Rizvi SM, Jager MJ, et al: In vitro targeting of NG2 antigen by 213Bi-9.2.27 alpha-immunoconjugate induces cytotoxicity in human uveal melanoma cells. Invest Ophthalmol Vis Sci 2005; 46(12): 4365-4371.
- Chekenya M, Rooprai HK, Davies D, et al: The NG2 chondroitin sulfate proteoglycan: role in malignant progression of human brain tumors. Int J Dev Neurosci 1999; 17(5-6): 421-435.
- Godal A, Bruland O, Haug E, et al: Unexpected expression of the 250 kD melanoma-associated antigen in human sarcoma cells. Br J Cancer 1986; 53(6): 839-841.
- Shoshan Y, Nishiyama A, Chang A, et al: Expression of oligodendrocyte progenitor cell antigens by gliomas: implications for the histogenesis of brain tumors. Proc Natl Acad Sci U S A 1999; 96(18): 10361-10366.
- Yadavilli S, Hwang EI, Packer RJ, Nazarian J: The Role of NG2 Proteoglycan in Glioma. Transl Oncol 2016; 9(1): 57-63.
- 97 Behm FG, Smith FO, Raimondi SC, et al: Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood 1996; 87(3): 1134-1139.
- Hilden JM, Smith FO, Frestedt JL, et al: MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood 1997; 89(10): 3801-3805.
- Schwartz S, Rieder H, Schlager B, et al: Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(-)/CD24(-)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia 2003; 17(8): 1589-1595.
- Smith FO, Rauch C, Williams DE, et al: The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996; 87(3): 1123-1133.
- Wuchter C, Harbott J, Schoch C, et al: Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia 2000; 14(7): 1232-1238.
- Wang X, Osada T, Wang Y, et al: CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst 2010; 102(19): 1496-1512.
- Nicolosi PA, Dallatomasina A, Perris R: Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015; 5(5): 530-544.
- de Vries JE, Keizer GD, te Velde AA, et al: Characterization of melanoma-associated surface antigens involved in the adhesion and motility of human melanoma cells. Int J Cancer 1986; 38(4): 465-473.
- Ozerdem U: Targeting pericytes diminishes neovascularization in orthotopic uveal melanoma in nerve/glial antigen 2 proteoglycan knockout mouse. Ophthalmic Res 2006; 38(5): 251-254.
- Ozerdem U: Targeting of pericytes diminishes neovascularization and lymphangiogenesis in prostate cancer. Prostate 2006; 66(3): 294-304.
- Ampofo E, Schmitt BM, Menger MD, Laschke MW: The regulatory mechanisms of NG2/CSPG4 expression. Cell Mol Biol Lett 2017; 22: 4.
- Stoiber S, Cadilha BL, Benmebarek MR, et al: Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019; 8(5).
- Sievers NM, Dörrie J, Schaft N: CARs: Beyond T Cells and T Cell-Derived Signaling Domains. Int J Mol Sci 2020; 21(10).
- Harrer DC, Dörrie J, Schaft N: Chimeric Antigen Receptors in Different Cell Types: New Vehicles Join the Race. Hum Gene Ther 2018; 29(5): 547-558.
- Montagner IM, Penna A, Fracasso G, et al: Anti-PSMA CAR-engineered NK-92 Cells: An Off-the-shelf Cell Therapy for Prostate Cancer. Cells 2020; 9(6).
- Van Cutsem E, Machiels J, Van den Eynde M, et al: SO-009 – Phase 1 studies assessing the safety and clinical activity of autologous and allogeneic NKG2D-based CAR-T therapy in metastatic colorectal cancer. Annals of Oncology 2019; 30: iv124-iv125.
- Santoro SP, Kim S, Motz GT, et al: T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res 2015; 3(1): 68-84.
- Bansal R, Reshef R: Revving the CAR – Combination strategies to enhance CAR T cell effectiveness. Blood Reviews 2020: 100695.
- Harrer DC, Dorrie J, Schaft N: CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy. Int J Mol Sci 2023; 24(3).
InFo ONKOLOGIE & HÄMATOLOGIE 2024; 12(1): 6–13