Drugs with a narrow therapeutic range, such as those frequently used in oncology, require a basic knowledge of possible interactions with other preparations. Pharmacodynamics and pharmacokinetics should not be foreign words.
Safe use of medications includes knowledge of potentially serious drug interactions. Especially in oncology, substances with a narrow therapeutic range are frequently used. Both subtherapeutic concentrations with risk for treatment failure and supratherapeutic concentrations with potential adverse effects want to be carefully avoided. A basic knowledge of the pharmacodynamics and pharmacokinetics of commonly used drugs – especially in the context of daily administered oral oncological therapeutics such as tyrosine kinase or checkpoint inhibitors – is a prerequisite to anticipate or recognize clinically relevant and adverse interactions. Management of drug interactions includes dose reduction, delayed application, or discontinuation of at least one of the interactants, depending on the benefit-risk assessment. In this educational article, an overview of relevant drug interactions is illustrated with selected clinical case examples and a specific focus on oncologic agents.
Pharmacological background
Safe use of medications includes knowledge of potentially serious drug interactions. Especially in oncology, compounds with a complex pharmacological profile, a steep dose-response/toxicity curve, pharmacokinetic and pharmacodynamic differences depending on the population (interpatient variability), and low therapeutic breadth are frequently used. All this means that the margin between therapeutic concentrations and potentially toxic drug levels is often small.
One “too many” can already lead to severe adverse effects. The same effects are present as in intoxication. Not the dose, but the concentration in the body makes the poison! All processes that lead to a corresponding increase can thus also result in symptoms of the increased effect – analogous to poisoning. Adverse interactions may even have fatal consequences as, for example, the combination of antiviral agents such as sorivudine with oral fluorouracil analogues (e.g. capecitabine) have shown.
A “too little may” mean a loss of activity, which may be associated with a worse prognosis. Especially here, the influencing factors are clinically difficult to grasp – differential diagnosis: nonresponder! A patient dies from his oncological disease earlier than the statistical mean. However, an administered inducer also leads to a reduction in chemo-associated side effects, which are mainly dose-dependent.
The multimorbid and polypharmaceutically treated patients already exhibit increased vulnerability to drug interactions. Other risk factors include organ dysfunction, especially renal insufficiency, use of enzyme inducers or inhibitors, and risk constellations with many involved physicians. The group of geriatric patients is particularly susceptible to interactions due to some peculiarities, in terms of pharmacokinetics (altered bioavailability, decreased clearance and organ functions, hypalbuminemia, reduction of extracellular volume, increase of adipose tissue fraction), polypharmacy and drug compliance. Especially in elderly patients, renal function based on calculated glomerular filtration rate (eGFR) may appear better than it is if immobility and reduced muscle mass result in low blood creatinine concentrations. Especially in the case of renally eliminated substances, a dose reduction is then recommended.
Drug interactions can in principle be divided into pharmacokinetic or pharmacodynamic interactions.
Pharmacokinetics describes the effect of the body on the substance – in terms of absorption, distribution, metabolism and excretion of a drug.
Pharmacokinetic interactions can occur at any level of ADME (absorption, distribution, metabolism, elimination), e.g..
Absorption: Interference of substances in the intestinal tract with complexation and consecutively reduced absorption.
Special focus – oncological substances and substance groups: Interactions with acid-suppressing drugs (proton pump inhibitors, H2 antagonists, antacids): Ex. Tyrosine kinase inhibitors: crizotinib, dasatinib, erlotinib, gefitinib, lapatinib, pazopanib. These combinations are best avoided or used with a time lag.
- Distribution: interference due to displacement from plasma protein binding with increase in volume of distribution with questionable clinical relevance. The unbound, free fraction of a drug is considered the pharmacologically active portion.
- Metabolism: Interference in metabolism with induction or inhibition of corresponding enzymes with consecutive accelerated or inhibited metabolism.
Specifically in focus – oncological substances and substance groups: An example of cytochrome (CYP) substrates would be the class of tyrosine kinase inhibitors (TKIs), all of which are metabolized to some extent via CYP3A4. Interactions with potent CYP3A4 inhibitors (eg, ketoconazole, voriconazole, itraconazole) and potent CYP3A4 inducers (eg. Rifampicin, enzalutamide) and TKIs: axitinib, crizotinib, dasatinib, erlotinib, gefitinib, imatinib, lapatinib, nilotinib, pazopanib, regorafenib, ruxolitinib, sunitinib, vemurafenib. Concurrent use should be avoided or accompanied by dose adjustments of the substrates (TKIs).
- Elimination: interference in elimination with competition for transport mechanisms with consequent delay in excretion and possible accumulation in the body.
Special focus – oncological substances and substance groups: Examples of renally eliminated oncological substances would be bendamustine, bleomucin, carboplatin, cisplatin, oxaliplatin, cyclophosphamide, methotrexate, pemetrexed, topotecan. Salicylates, probenecid, or sulfonamides may compete for renal tubular secretion when used concomitantly with methotrexate, resulting in reduced elimination and consequent increase in concentration of methotrexate.
Pharmacokinetic interactions lead to a change in the plasma level of the substances, which may result in a decreased or an increased effect. Representatives of the cytochrome P450 enzyme family, drug transporters such as P-glycoprotein or organic ion transporters are often involved. Table 1 shows example substances of frequently used inducers and inhibitors of CYP3A4.
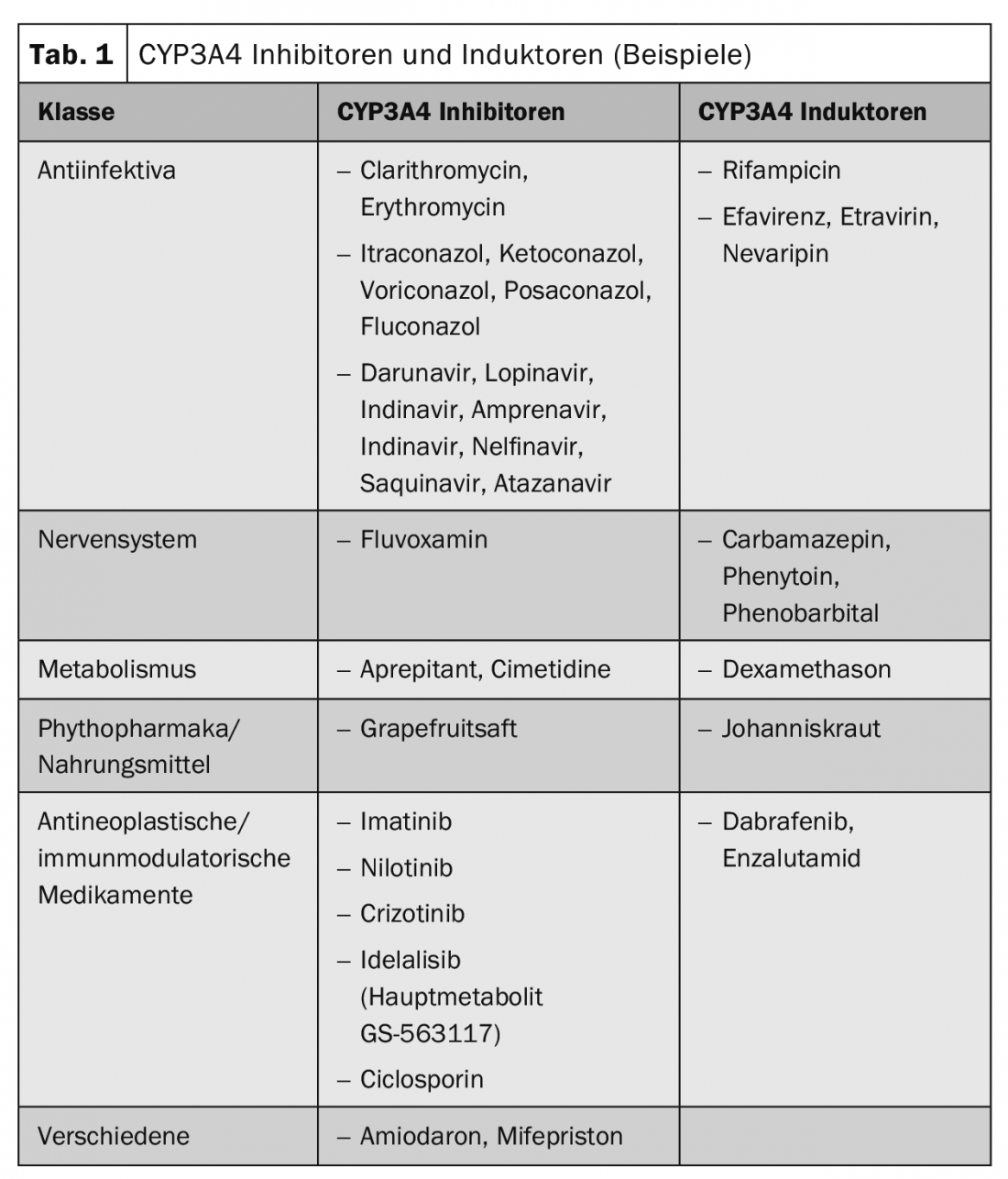
Special focus – oncological substances and substance groups: Tamoxifen is itself a prodrug that is activated via CYP2D6. Particular caution is therefore required with inhibitors of this isoenzyme. Tamoxifen is metabolized to its active metabolite endoxifen with the involvement of CYP2D6. In addition, CYP3A4 and, to a lesser extent, CYP2C9 and CYP2C19 are involved in the metabolism of tamoxifen. Inhibitors of CYP2D6 may therefore affect the metabolism of tamoxifen in a clinically relevant manner, resulting in reduced efficacy or loss of efficacy of tamoxifen. Concomitant administration of CYP2D6 inhibitors such as paroxetine resulted in up to 75% reduction in plasma concentrations of the active metabolite endoxifen in published studies. Concomitant use of CYP2D6 inhibitors (such as paroxetine, fluoxetine) together with tamoxifen should therefore be avoided. The antidepressant venlafaxine, on the other hand, would not be a CYP2D6 inhibitor.
Conversely, oncology drugs themselves may not only be substrates for CYP enzymes, but also inhibitors or inducers of isoenzymes.
Specifically in focus – oncology substances and substance groups: Idelalisib, a phosphatidylinositol 3-kinase inhibitor, is converted to the major metabolite GS-563117, which is a potent CYP3A inhibitor. The CYP3A4 marker reaction with midazolam showed a midazolam increase in Cmax of 140% and AUC of 440%. Therefore, there is also the possibility of interaction with other drugs that are metabolized via CYP3A4, such as. Ciclosporin, sirolimus, tacrolimus, amlodipine, carbamazepine, colchicine, diltiazem, felodipine, fentanyl, ketoconazole, itraconazole, posaconazole, voriconazole, lidocaine, methadone, nifedipine, nicardipine, trazodone, warfarin, phenprocoumon, boceprevir, telaprevir, clarithromycin, telithromycin, atorvastatin, tadalafil, buspirone, clorazepate, diazepam, estazolam, flurazepam, zolpidem, or tyrosine kinase inhibitors such as erlotinib. A combination with idelalisib, for example, necessitates a reduction of the erlotinib dose, which should then be lowered in steps of 50 mg according to the SmPC. The recommended dose of erlotinib is usually 150 mg once daily at least one hour before or two hours after eating.
Other degradation or remodeling pathways independent of cytochrome P450 are also known. Again, some clinically relevant interactions are possible.
Specifically in focus – oncological substances and substance groups: Fluorouracil is metabolized mainly in the liver to inactive products, including carbon dioxide, urea, and α-fluoro-β-alanine (FBAL). The enzyme dihydropyrimidine dehydrogenase (DPD), for which a genetic polymorphism is known, is involved in metabolism. Fluorouracil metabolism is slowed in patients with dihydropyrimidine dehydrogenase (DPD) insufficiency. This is also the subject of a current procedure of the European Medicines Agency (EMA), in which existing screening methods for DPD deficiency are being investigated. However, certain substances can also irreversibly inhibit DPD. Drugs that contain fluorouracil, such as Capecitabine or Tegafur (prodrugs of 5-FU), must not be used together with brivudine or chemically related substances such as sorivudine. Enzyme inhibition can lead to increased toxicity of 5-FU products, which can be potentially fatal. There should be at least a 4-week interval between treatment with brivudine, sorivudine, or analogues and initiation of therapy with 5-FU products.
Pharmacodynamics, on the other hand, describes the effect of the substance on the body.
In this type of interaction, the pharmacological activity of the respective substance is influenced without changing the concentration at the site of action by combination with a substance with a similar or opposite effect, resulting either in an enhancement or attenuation of the therapeutic effect. This does not change the concentrations in the blood. Therapeutic drug monitoring (TDM) with plasma level determinations therefore does not provide any additional information in this case. For example, various substances, as well as classical oncological agents (e.g., anthracyclines), can prolong QTc time. These ECG changes are also commonly reported for tyrosine kinase inhibitors. The potential depends on the chemical structure and the plasma concentration of the substance. Therefore, QTc time prolongation can be further enhanced in combination with CYP inhibitors but also the simultaneous use of different drugs that affect QTc time.
Specifically in focus – oncological substances and substance groups: Examples of tyrosine kinase inhibitors that interact with QTc-time prolonging agents: Crizotinib, gefitinib, lapatrinib, nilotinib, pazopanib, sorafenib, sunitinib, vandetanib, vemurafenib. If specific combinations are clinically necessary, ECG checks are recommended before therapy is started and then as it progresses.
Substance-drug (“drug-drug”) interactions are of particular importance in this context, as they may be predictable from prior case reports, clinical trials, or understanding of pharmacologic principles. Adverse drug interactions can have life-threatening consequences, which can lead to established substances also having to be withdrawn from the market.
Pharmaceutical interactions as physicochemical incompatibilities of two substances outside the body can lead to complexation, degradation or precipitation. However, these are not the subject of this overview.
Besides drug-drug interactions, there are also drug-herb interactions (e.g., with ginkgo, ginseng, garlic), drug-food interactions (e.g., with milk, grapefruit juice), drug-alcohol interactions (e.g., with benzodiazepines and many other sedating substances), or drug-disease interactions (e.g., metoclopramide and Parkinson’s disease). Interactions with other substances and exogenous substances, such as tobacco, are also possible. Tobacco ingredients cause CYP1A1 and CYP1A2 induction in smokers.
Special focus – oncological substances and substance groups: Case study tobacco and erlotinib: For example, erlotinib is metabolized in the liver via the isoenzymes CYP3A4 and CYP1A2 and in the lung possibly via CYP1A1. This results in a 50-60% reduction in erlotinib exposure in smokers. In one pharmacokinetic study, exposure was one-half to one-third of that found in nonsmokers or ex-smokers as a result of induction of CYP1A1 in the lung and CYP1A2 in the liver. For this reason, smoking cessation should additionally be recommended to smokers.
On the other hand, interactions, especially in oncology with many combination therapies, are also desirable. However, depending on the study, drug interactions are also responsible for 20-30% of adverse drug reactions, the majority of which become clinically relevant. As the number of drugs administered increases, so does the potential for adverse drug interactions. They are associated with increased morbidity and mortality, prolonged hospitalization of affected patients, and increased health care costs. While some of the adverse drug interactions and side effects cannot be predicted, many can be anticipated and prevented.
Cases from practice
Pharmacokinetic interaction: chemotherapy and antiretroviral therapy: a 63-year-old man with long-standing HIV infection with appropriate antiretroviral therapy and coronary 3-vessel disease presents with newly indistinct masses in the pancreatic head. In V.a. pancreatic head carcinoma, chemotherapy with gemcitabine and nab-paclitaxel should be evaluated for interactions with existing therapy (Table 2).
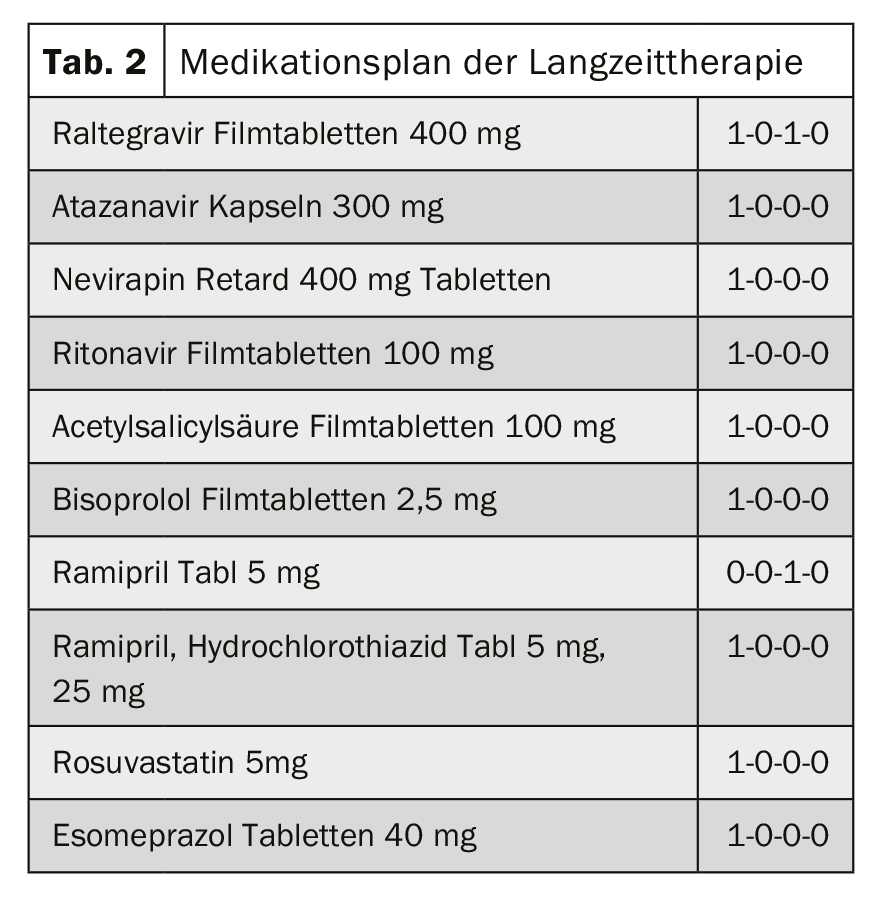
Assessment and Background: Nab-paclitaxel is an antimicrotubule agent. Nab-paclitaxel is metabolized via CYP2C8 and CYP3A4. Drugs that induce or inhibit the CYP enzyme system (CYP2C8 and/or CYP3A4) may therefore affect the plasma levels of nab-paclitaxel and thus its efficacy.
- Interaction Nab-paclitaxel and ritonavir: Ritonavir is a potent inhibitor of CYP2C8 and CYP3A4. The strong inhibition of these enzymes may also lead to increased plasma levels of nab-paclitaxel.
- Interaction Paclitaxel and Atazanavir: Atazanivir is a strong inhibitor of CYP3A4 and a weak inhibitor of CYP2C8. Strong inhibition of CYP3A4 may result in increased plasma levels of nab-paclitaxel. The weak inhibition of CYP2C8 is likely to play a minor role – but could add up with other interactions. Although these interactions have not been formally substantiated by pharmacokinetic studies, an enhanced effect and increased dose-dependent risk of side effects can be assumed based on theoretical derivations.
Based on theoretical derivations, pharmacokinetic interactions of atazanavir and ritonavir with nab-paclitaxel lead to increased plasma levels of the antimicrotubule agent. With combination therapy, concentration-dependent adverse drug effects of nab-paclitaxel would be expected, such as bone marrow depression, mucositis, and peripheral neuropathy. Due to the narrow therapeutic range of chemotherapy, myelotoxicity as a potentially life-threatening complication, and multiple degradation pathways of paclitaxel that would be inhibited, switching the combination partners atazanavir/ritonavir and Nab-paclitaxel is recommended.
The pharmacodynamic interaction: checkpoint inhibitor & “immunomodulators”/immunosuppressants [7]: A 53-year-old female patient with Z.n. liver transplantation 36 months ago due to hepatocellular carcinoma based on liver cirrhosis with chronic hepatitis C infection, receives prednisone, mycophenolate and everolimus as immunosuppressive therapy. Due to HCC recurrence with metastases two years after transplantation, sorafenib is administered. Due to side effects and also progression of the underlying disease, this is stopped again after two months. Nivolumab at a dose of 200 mg (3 mg/kg body weight) is started. As a result, liver function parameters increase after one week. A liver biopsy is performed, which shows signs of acute cellular rejection. It is then treated with tacrolimus as well as methylprednisone 500 mg/day. There is further deterioration of liver function, clinical condition, CNS depression in intracerebral hemorrhage with intensive care and eventual fatal outcome.
Assessment and Background: Immune checkpoint inhibitors such as nivolumab have revolutionized the treatment of various tumor diseases. Nivolumab (Opdivo®) is a human IgG4 monoclonal antibody that blocks PD-1 receptors on activated T cells, B cells and natural killer T cells, monocytes, and dentritic cells. PD-1 receptors have 2 ligands, PD-L1 and PD-L2, which are expressed on tumor cells and antigen-presenting cells. Binding of the receptor to ligands leads to negative regulation of T cells and prevention of T cell response. Preventing binding leads to a T-cell response and anti-tumor effect. On the other hand, immune tolerance in organ transplantation is essential for this patient. PD-1 and CTLA-4 signal transduction lead to immune tolerance. Therefore, inhibition may trigger immune-mediated organ failure in these patients.
According to the Swiss SmPC, “treatment with nivolumab or nivolumab in combination with ipilimumab should not be resumed while the patient is receiving immunosuppressive corticosteroid doses or other immunosuppressive therapy.” Also, “the use of systemic corticosteroids and other immunosuppressants should be avoided before starting treatment with nivolumab because of their potential interference with pharmacodynamic activity.” Based on pharmacodynamic considerations, this interaction may become relevant in both directions: A reduction in the immunosuppressive effect of drugs important after transplantation, but also a reduction in the anti-tumor effect of checkpoint inhibitors.
Clinical approach to drug interactions
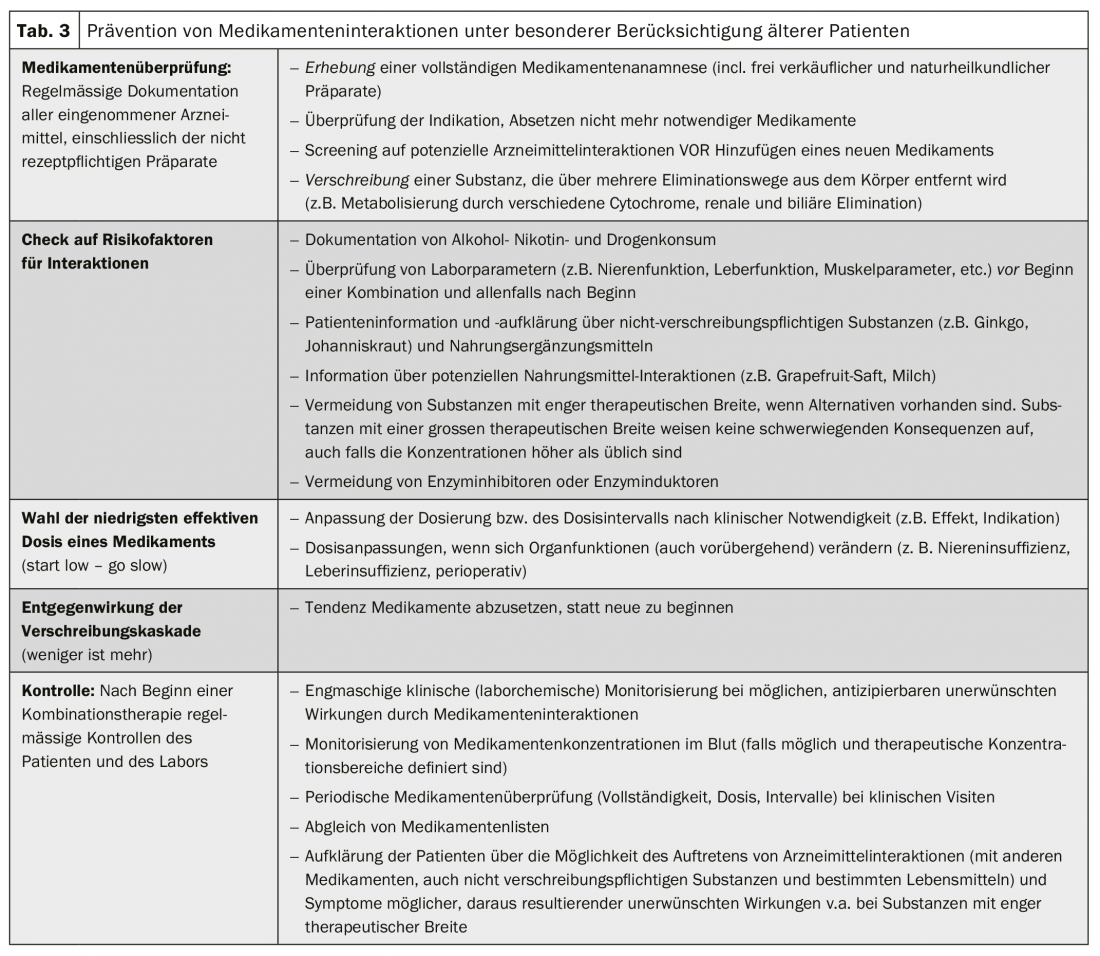
Drug interactions can cause clinical problems to varying degrees. Ideally, these are anticipated before they occur. Particularly in the case of polypharmacy, certain rules should be observed (Tab. 3). Unrecognized interactions and side effects can also end in a so-called prescription cascade: Side effects are not recognized as such and new therapies are administered against the new “symptoms” instead of recognizing the pharmacological trigger. When interactions with adverse effects occur, management consists of discontinuation or dose reduction of the suspected substance(s), continued monitoring, clinical controls, or therapeutic level measurements (Tab. 4). The detection and prevention of adverse, clinically relevant drug interactions is a team effort and involves not only the patient but also all involved physicians, nurses and pharmacists.

It was shown that interaction-related hospitalizations could be avoided by adequate controls and rational selection of the most appropriate medication in the situation. Assistance with drug interactions is provided either by current textbooks, interaction programs or interaction websites (box). However, computer programs designed to help detect dangerous combinations fail in up to one-third of interactions, while on the other hand they often unnecessarily warn of trivial and clinically irrelevant interactions. It is therefore often useful to survey more than one interaction database.
|
Web pages for interaction checks
|
Principled considerations of potential interaction provide further support:
- Is the medication list complete, incl. herbal preparations?
- Are there any substances among them that are particularly susceptible to interaction?
- Are there potential alternatives to these substances?
- How can the effects be monitored?
- Is this a pharmacokinetic interaction where therapeutic drug monitoring with level determinations might be helpful?
- What is the severity of the adverse effect? How great is the clinical relevance of the therapy? (risk-benefit analysis)
- Is the interaction dose or concentration dependent? Can an improvement in symptoms be expected with a dose reduction?
- Are other patient-specific factors present that promote the adverse reaction (e.g., renal insufficiency)? Can these be optimized?
Summary
In summary, it is important to think about potential drug interactions in the first place. This is particularly relevant when a new drug is added to an already complex therapeutic regimen. New oncological substances, such as targeted therapies, are often taken daily. Therapeutic breadth is often narrow. Oncologic patients who are elderly and have impaired elimination capacity-such as renal dysfunction-are particularly susceptible to adverse effects due to drug-drug interactions.
Take-Home Messages
- In oncology in particular, substances with a narrow therapeutic range are predominantly used.
- Before adding a new substance (drug but also phythopharmaceutical) to an already existing therapy plan, it should be checked for potential interactions.
- Special interaction programs, interaction websites with different precision are available for drug interaction checks.
- Options for adverse drug interactions vary depending on the type and severity or risk-benefit ratio. Dose reduction, change in application time, or even discontinuation of the suspected substance(s) may be necessary.
- In the case of suspected or ineluctable drug interactions, close clinical or laboratory monitoring and, if necessary, measurements of drug concentrations in the blood (in the case of pharmacokinetic interactions) are useful.
Literature:
- Wilkinson GR: Drug Metabolism and Variability among Patients in Drug Response. N Engl J Med 2005; 352: 2211-2221.
- van Leeuwen RW, et al: Drug-drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol 2014; 15: e315-e326.
- van Leeuwen RW, et al: Prevalence of potential drug-drug interactions in cancer patients treated with oral anticancer drugs. Br J Cancer 2013; 108: 1071-1078.
- Mallet L, Spinewine A, Huang A: The challenge of managing drug interactions in elderly people. Lancet. 2007 Jul 14;370(9582): 185-191.
- Beijnen JH, Schellens JHM: Drug interactions in oncology. Lancet Oncol. 2004 Aug;5(8): 489-496.
- Van Meerten E, Verweij J, Schellens JHM: Antineoplastic agents: drug interactions of clinical significance. Drug Safety 1995; 12: 168-182.
- Gassmann D, et al: Liver Allograft Failure After Nivolumab Treatment-A Case Report With Systematic Literature Research. Transplant Direct. 2018 Jul 20;4(8): e376.
- Weiler S, et al. Tips for practice: clinically relevant adverse drug interactions. Switzerland Med Forum 2015. 15: 152-156.
InFo ONCOLOGY & HEMATOLOGY 2019; 7(2-3): 5-10.