In the past, a diagnosis of cystic fibrosis (CF) was synonymous with early death usually before the age of 30. The hereditary disease is still life-shortening, but thanks to advances in research and new therapies, average life expectancy in Central Europe has already been increased to over 50 years. For those affected in adulthood, there are now a number of options for managing cystic fibrosis.
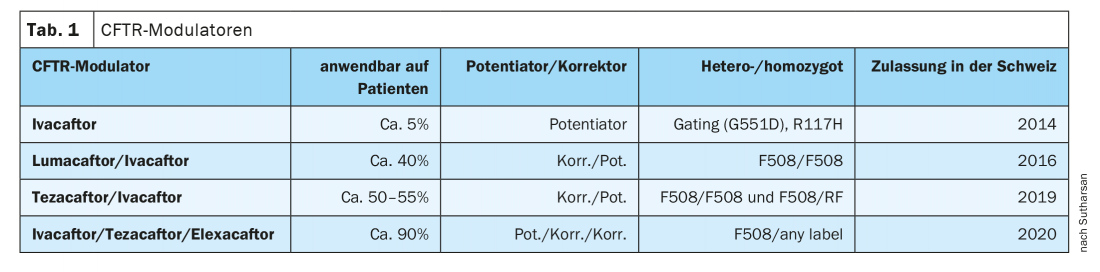
The introduction of pancreatic enzymes has been a major therapeutic advance, but the establishment of centers where CF patients receive targeted treatment has also helped to increase the likelihood of survival, said Dr. Sivagurunathan Sutharsan, senior physician at the Department of Pneumology, Ruhrland Clinic, University Medical Center Essen (D), by way of introduction. In 1989, the CFTR gene was discovered. Although no gene therapy exists to date, personalized medicine has also arrived in cystic fibrosis, the expert said: in 2014, ivacaftor (IVA) was approved in Switzerland as the first active ingredient for a small group of patients (5-6%); in the meantime, there are three other modulators that are available as combinations and, since August 2020, also as a triple (Tab.1).
Increasing complications with increasing age
The first complications appear in infancy: infants frequently (up to 25%) present with meconium ileus as the earliest indicator, usually (>85%), pancreatic insufficiency is already present and lung infections often already occur (but only <25% Pseudomonas aeruginosa). In the course of childhood and adolescence, the proportion of P. aeruginosa increases to approx. 45%, but other organs also come into play, including liver disease and nasal polyps (25%). In adulthood, the full-blown picture finally emerges, including severe lung and bone diseases (overview 1).
During the first decade of life, Staphylococcus aureus and Haemophilus influenzae are the most common bacteria isolated from sputum in CF; in the second and third decades of life, Pseudomonas aerugiosa is much more common.

Exacerbations increase the risk
Central to the management of cystic fibrosis is avoiding exacerbations. “Patients who have more than 2 pulmonary exacerbations per year are at increased risk for death or lung transplantation,” Dr. Sutharsan said. Reducing the number of exacerbations and subsequent lung function loss should therefore be a major goal of CF management.
In management, physiotherapy is usually prescribed to get the secretions out of the lungs; antibiotic therapy is indicated for chronic infections with Pseudomonas or Staphylococcus, and macrolides such as azithromycin are often used as continuous therapy for chronic Pseudomonas colonization. If patients are bleeding, one has the option of performing bronchial artery occlusion. Therapy with CFTR modulators is new (overview 2).

CFTR modulators
In the most common mutation, F508del, the protein is produced but it is usually degraded in the cell. But with the new modulators, the CFTR correctors elexacaftor (ELX) and tezacaftor (TEZ), the chloride channel is stabilized at different binding sites, preventing degradation. Directed transport to the cell surface follows, and there the CFTR potentiator ivacaftor can increase the opening probability of the chloride channel. There are now 4 approved CFTR modulators. While ivacaftor can be used in about 5% of the CF population, the new triple combination of ivacaftor/tezacaftor/elexacaftor can treat about 90% of patients worldwide. As a prerequisite for treatment, one must have at least one 508 mutation on one allele. “The second mutation usually doesn’t matter,” Dr. Sutharsan said. Side effects include rash and itching, which are usually well controlled with oral antihistamines or steroids. However, a good effect is described not only on the lungs, but also on the sinuses.
In his Essen clinic, Dr. Sutharsan and his colleagues have matched 133 CF patients to the new triple ELX/TEZ/IVA (80 homozygous, 32 + 21 (hardship) heterozygous). In the homozygous group, there were 96 hospitalizations with i.v. therapy due to exacerbations in the 12 previous months. In 6 months under the triple, this number reduced to only 5 i.v. therapies. The situation was similar in the heterozygotes: 29 + 74 (hardship) i.v. treatments before initiation over a 12-month period became only 2 + 7 (hardship) i.v. treatments in the first 6 months under ELX/TEZ/IVA. While further progress remains to be seen, Dr. Sutharsan concluded, the figures “already show the enormous therapeutic progress that the new modulators are bringing.”
Source: 128. Congress of the German Society for Internal Medicine (DGIM)
InFo PNEUMOLOGY & ALLERGOLOGY 2022; 4(2): 30-31.