Early and adequate treatment can have a positive impact on the further course of this rare multisystem disease. This article provides a brief overview of what is known today. Among other things, the identification of new biomarkers could serve as the basis for personalized therapy in the future.
Juvenile dermatomyositis (JDM) is a systemic, immune-mediated disease of childhood characterized by muscle weakness and typical skin rashes [9]. Other organ systems, such as the heart and lungs, may also be affected and are still the most common cause of death in these patients. It is the most common idiopathic inflammatory myopathy in childhood, with an incidence of 2-4/million/year.
The inflammatory process is characterized by infiltration of the affected tissue by cells of the immune system, such as T- cells and plasmacytoid dendritic cells, and an interferon signature [1]. Underlying most of the symptoms is a vasculopathy, which results in a lack of functioning and loss of endothelial cells. Effective immunosuppression, usually a combination therapy of subcutaneous methotrexate and oral corticosteroids, has greatly reduced mortality (currently at approximately 1%) and approximately 50% of patients show only one disease flare with therapy before remission can be achieved [2]. The other 50% show a chronic course, with frequent relapses, increased morbidity, and reduced quality of life [2].

Subphenotyping by biomarkers
JDM is a very heterogeneous disease with different phenotypes, and diverse organ systems may be affected to varying degrees. Autoantibodies have been the target of intense research in recent years and have better characterized the diverse phenotypes; therefore, autoantibodies have prognostic value [3]. Monitoring disease activity and organ damage caused by inflammation is an important part of the management of these young patients, as it determines the long-term outcome of this disease [4]. Various tools are available to the clinician in this regard, such as histopathology of muscle and skin tissue, capillary microscopy (Fig. 1), clinical scores to assess muscle and skin involvement [5], and now various biomarkers in the blood [6,7]). The clinical scores used to assess disease activity require patient cooperation, which is sometimes challenging in younger children. Also, these clinical scores are of limited use in detecting subthreshold inflammation or in differentiating other causes of muscle weakness such as steroid toxicity.

Guidelines for optimizing disease management
In 2012, the European initiative SHARE (Single Hub and Access point for paediatric Rheumatology in Europe) was launched with the aim of standardizing diagnostic and therapeutic management for children and adolescents with rheumatic diseases across Europe. For JDM, these recommendations were published in 2017 [6].
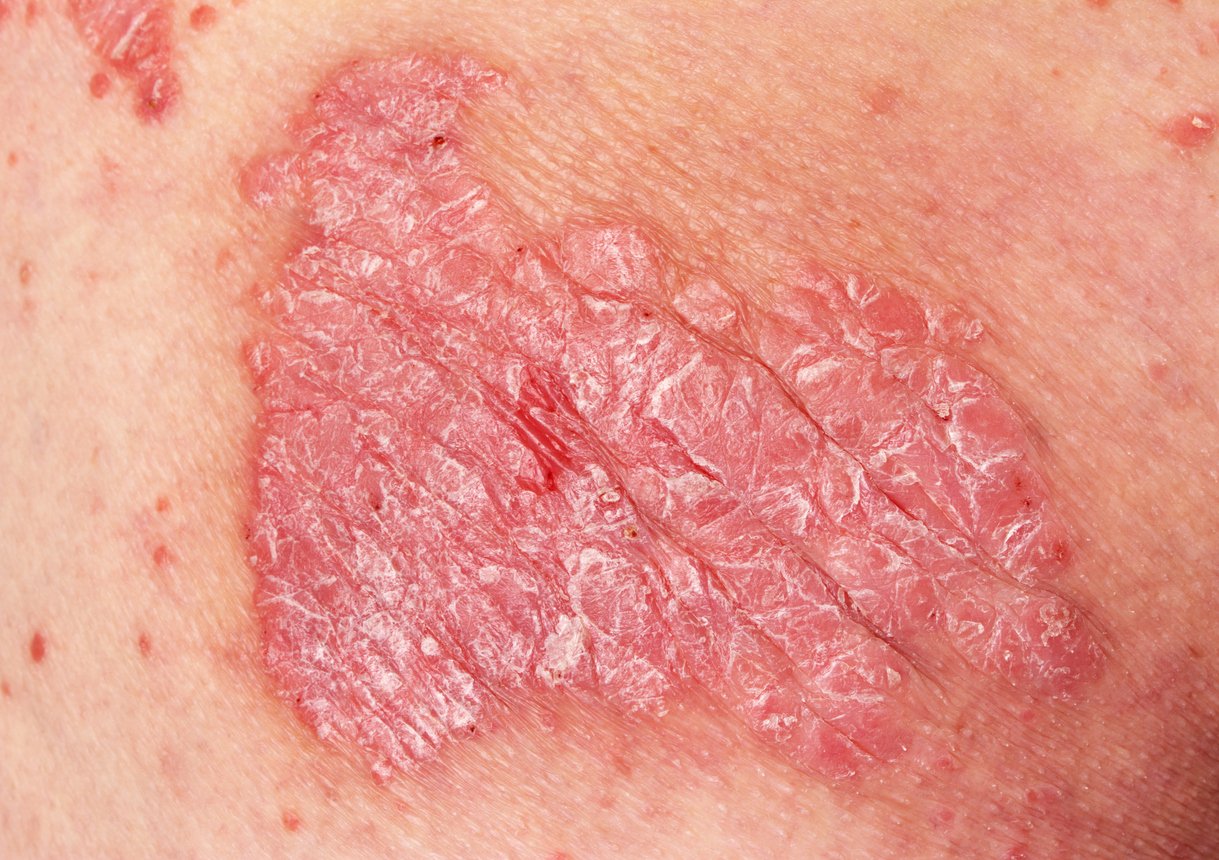
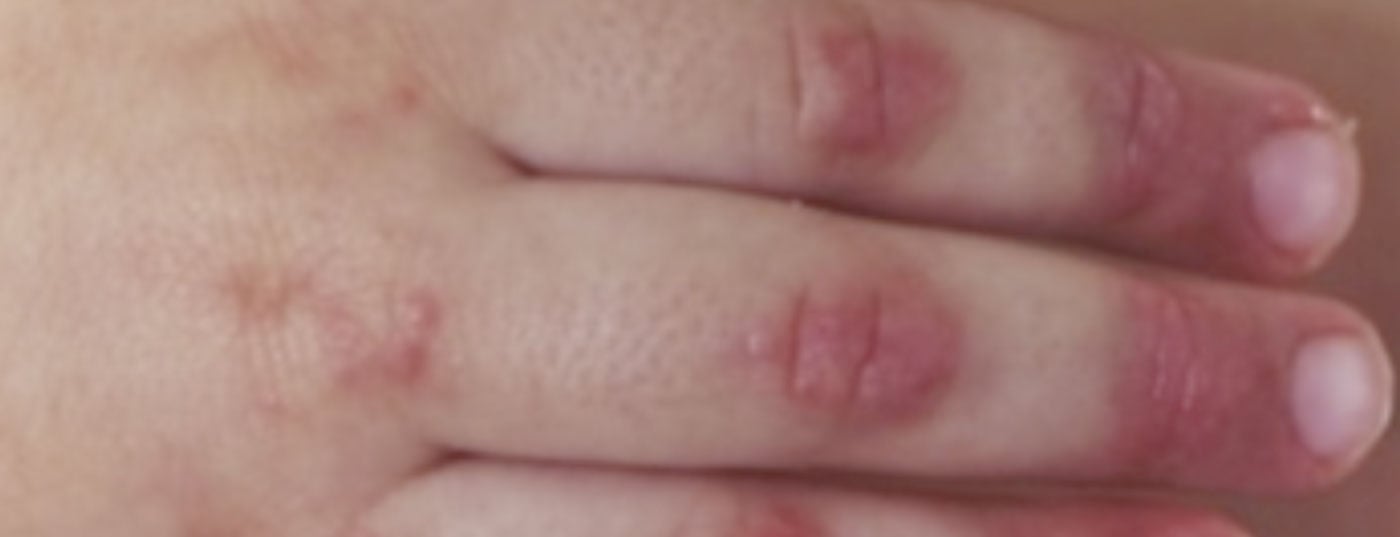
Diagnosis: The diagnosis of juvenile dermatomyositis remains primarily clinical with typical skin findings (Fig. 2) and proximally emphasized muscle weakness. Therefore, if JDM is suspected, rapid referral to a nearby center of excellence should be made. Early and adequate therapy can prevent long-term damage and increase the chance of early remission. Bohan and Peter’s 1975 classic diagnostic criteria include five elements:
- characteristic skin symptoms
- Proximal muscle weakness
- typical pathological muscle biopsy
- Increased muscle enzymes
- myopathic changes in the electromyogram (EMG)
→ In pediatric rheumatology practice, EMG has been replaced by nonpainful MRI (Fig. 3) [8].
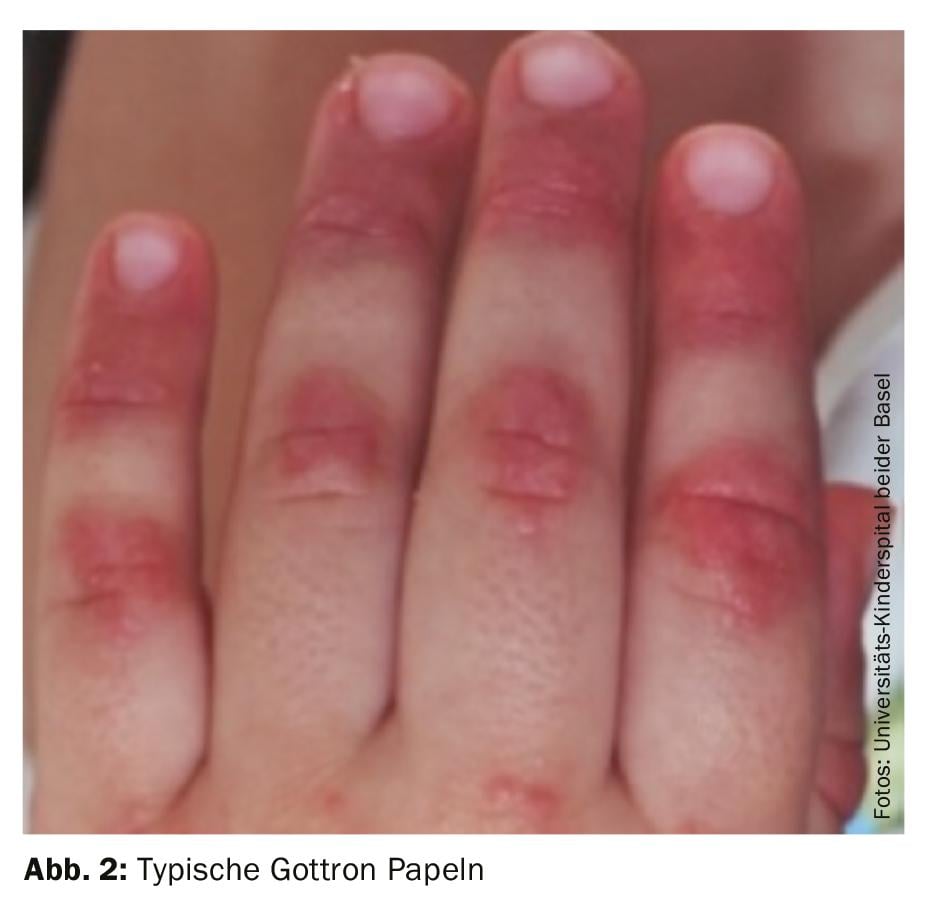

Newer classification criteria were published in 2017; for example, they omit muscle biopsy for pathognomonic skin symptoms and also include autoantibodies (anti-Jo-1) in the criteria [9]. Ultrasound is also being used more frequently because it does not require sedation in younger children.
Muscle weakness should be assessed in the child using a variety of instruments. Functional muscle tests, called the Children’s Myositis Activity Score (CMAS) [10] as well as muscle tests that measure strength alone, called the Manual Muscle Test (MMT) [11] are available. Both tests are helpful in assessing muscle weakness at diagnosis; however, they also serve as important follow-up parameters that can provide a significant indication of disease activity. Both tests are part of the 2013 remission criteria [12].
Wienke et al. recently validated several biomarkers (galectin-9 and CXCL10) that play a role not only in diagnosis but also in follow-up and may also facilitate treatment decisions at the onset of the disease: In this prospective study, patients with high levels of these biomarkers at the time of diagnosis showed a more severe course in the follow-up and could therefore potentially benefit from more intensive therapy. In contrast, in patients with only one relapse, the values had almost normalized under therapy already a few months after diagnosis. Furthermore, it has been shown that the markers showed a renewed increase in blood levels even before the clinical findings of a disease flare [7].

2nd therapy: First-line therapy remains a combination of steroids and me-thotrexate, possibly ciclosporin [6]. Methotrexate should be maintained for at least 2 years, and steroids are discontinued in the 1st year of therapy if the course is single-phase [6]. If certain symptoms such as severe muscle weakness (child cannot get out of bed, severe dysphagia), impaired myocardial or pulmonary function, or skin ulcers indicate a severe course, cyclophosphamide should be added. In the absence of improvement, intravenous immunoglobulins or ciclosporin A should be added or switched to biologics (rituximab, infliximab, adalimumab). From a pediatric perspective, increasing calcinosis of subcutaneous or muscle tissue (Figs. 3 and 4) is a sign of persistent systemic inflammation that warrants intensification of therapy. Stem cell transplantation has also been successfully performed in JDM patients with a very severe course; some patients achieved complete remission without therapy. When clinical remission is achieved, calcinosis may regress completely.
With good success, this standard therapy remains associated with the risk of adverse side effects, such as infection, steroid-induced muscle weakness, or weekly nausea with methotrexate. In the future, the aforementioned new biomarkers could enable the early identification of a new disease flare or a more rapid reduction of therapy. The biomarkers could thus support the path to personalized therapy for this rare disease.
| Web-based diagnostic tool Meanwhile, a web tool is available that gives a probability of diagnosis with the classification of the subgroup in addition to the diagnosis. These criteria have been validated for both children and adults with suspected inflammatory myopathy. http://www.imm.ki.se/biostatistics/calculators/iim/ |
Literature:
- Piper CJM, et al: CD19(+)CD24(hi)CD38(hi) B Cells Are Expanded in Juvenile Dermatomyositis and Exhibit a Pro-Inflammatory Phenotype After Activation Through Toll-Like Receptor 7 and Interferon-alpha. Front Immunol 2018; 9: 1372.
- Gowdie PJ, Allen RC, Kornberg AJ, Akikusa JD: Clinical features and disease course of patients with juvenile dermatomyositis. Int J Rheum Dis 2013; 16(5): 561-567.
- Rider LG, Nistala K: The juvenile idiopathic inflammatory myopathies: pathogenesis, clinical and autoantibody phenotypes, and outcomes. J Intern Med 2016; 280(1): 24-38.
- Huber A, Feldman BM: Long-term outcomes in juvenile dermatomyositis: how did we get here and where are we going? Curr Rheumatol Rep 2005; 7(6): 441-446.
- Huber AM, et al: Preliminary validation and clinical meaning of the Cutaneous Assessment Tool in juvenile dermatomyositis. Arthritis Rheum 2008; 59(2): 214-221.
- Enders FB, et al: Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis 2017; 76(2): 329-340.
- Wienke J, et al: Galectin-9 and CXCL10 as Biomarkers for Disease Activity in Juvenile Dermatomyositis: A Longitudinal Cohort Study and Multicohort Validation. Arthritis Rheumatol 2019. doi.org/10.1002/art.40881
- Brown VE, et al: An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford) 2006; 45(8): 990-993.
- Lundberg IE, et al: 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis 2017; 76(12): 1955-1964.
- Huber AM, et al: Validation and clinical significance of the Childhood Myositis Assessment Scale for assessment of muscle function in the juvenile idiopathic inflammatory myopathies. Arthritis Rheum 2004; 50(5): 1595-1603.
- Jain M, et al: Intra-rater and inter-rater reliability of the 10-point Manual Muscle Test (MMT) of strength in children with juvenile idiopathic inflammatory myopathies (JIIM). Phys Occup Ther Pediatr 2006; 26(3): 5-17.
- Lazarevic D, et al: The PRINTO criteria for clinically inactive disease in juvenile dermatomyositis. Ann Rheum Dis 2013; 72(5): 686-693.
- Mathiesen PR, et al: Aerobic fitness after JDM–a long-term follow-up study. Rheumatology (Oxford) 2013; 52(2): 287-295.
DERMATOLOGIE PRAXIS 2019; 29(4): 26-29