
The therapeutic focus remains on symptomatic treatment options and the avoidance of provoking factors. Molecular, potentially curative therapeutic options for epidermolysis bullosa are still in the research stage. In the diagnostic workup of this geno- and phenotypically heterogeneous autoimmune disease, there are several things to keep in mind.
Epidermolysis Bullosa (EB) subsumes a group of geno- and phenotypically heterogeneous genodermatoses characterized by an exaggerated mechanical fragility of epithelialized tissues. With a prevalence of approximately 500,000 cases worldwide, EB is a rare disease [1,2]. To date, mutations have been described in over 20 genes encoding components involved in the assembly of cytoskeletal keratin filaments, adhesion molecules, desmosomes, hemidesmosomes, and anchoring fibrils of epithelia (Fig. 1). Consequently, the structural and functional integrity of intraepidermal adhesion and dermoepidermal adherence to skin and mucous membranes is impaired, affecting barrier function, cell-cell and cell-matrix interaction, proliferation, tissue regeneration, and differentiation processes [3–5]. The combinatorial spectrum of type, homo- or heterozygosity, number (mono- or digenetic inheritance) and localization of the mutation in the respective gene segment, as well as the resulting quantitative (absence or reduction) and qualitative (gradual loss of function) disturbance of protein expression, causes considerable geno- and consequently phenotypic variances of EB. In addition to the primary genetic defect, secondary epigenetic and biochemical factors (e.g., induction of chronic inflammatory cascades and “tissue remodeling”), as well as environmental factors, also influence clinical severity [6,7].
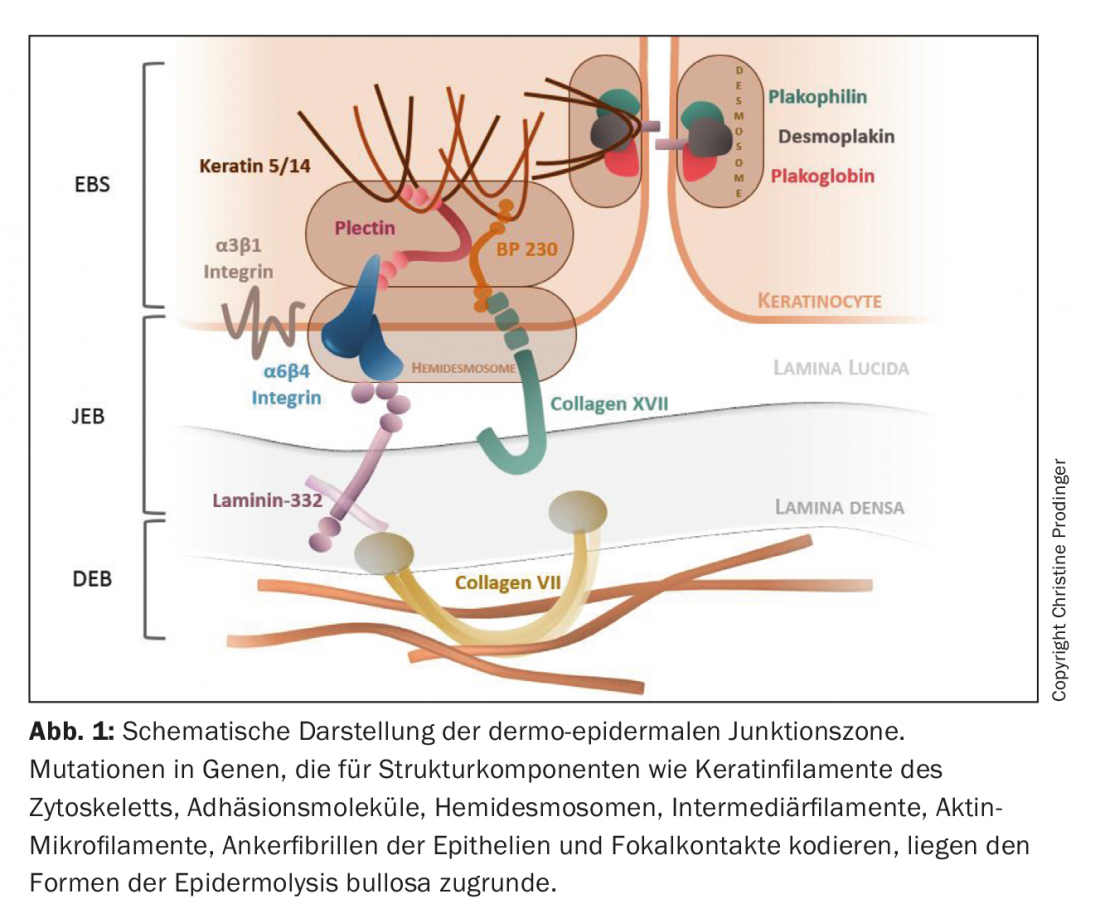
Since the affected index genes are not only expressed in skin and skin-related mucous membranes, but additionally in other epithelia (respiratory, genitourinary and gastrointestinal tract) and in mesenchymal tissue (skeletal muscle), primary extracutaneous manifestations are also possible there. Thus, EB can develop into a systemic disease with significant morbidity and mortality [8].
Pathophysiological classification
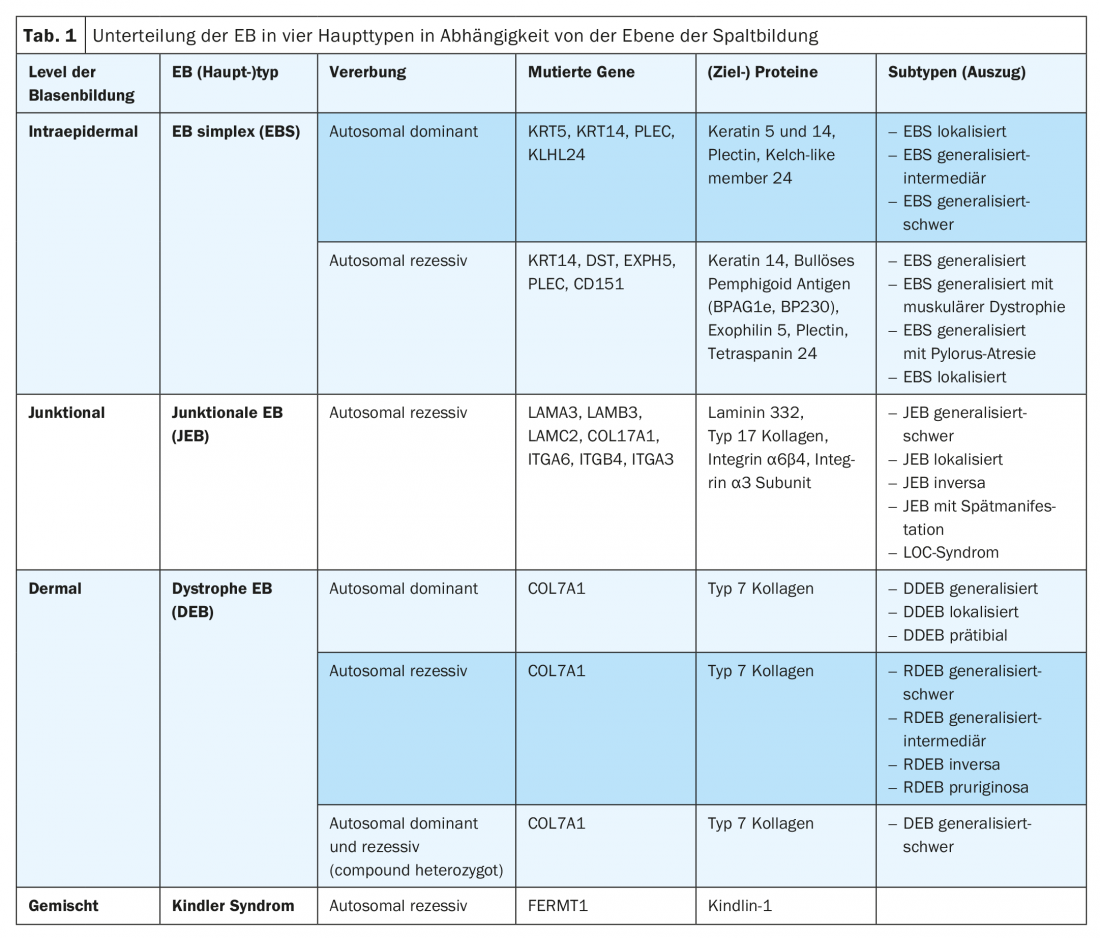
The leading symptom of EB is the subtype-dependent variable, increased to excessive fragility of the skin and mucous membranes to mechanical stress with formation of blisters, erosions, ulcerations, crusts or scars. According to the level of cleavage as a function and consequence of the underlying mutation, EB is divided into four main types (Table 1) [9]. Due to the increasing availability of modern molecular diagnostic methods (e.g. “next generation sequencing”), new EB (sub)types are also being described again and again. Thus, a mutation in the KLHL24 gene encoding a component of the ubiquitin ligase complex could be assigned to an autosomal dominant variant of EB simplex, resulting in excessive ubiquitination and degradation of keratin 14 [10–12]. In a Kindler syndrome-like phenotype, a mutation was also recently identified in the CD151 gene, which encodes a tetraspanin in the basement membrane zone. This transmembrane protein interacts with integrins and is involved in cellular growth, development, and motility processes [13]. In addition, a mutation was recently discovered in the PLOD3 gene, which encodes lysyl hydroxylase 3 and regulates posttranslational processing of type 7 collagen. Clinically, affected individuals show extensive connective tissue defects, joint contractures, skeletal malformations, and growth sum retardation. The level of bubble formation is similar to recessive dystrophic EB in the sublamina densa [14].
Diagnostic algorithm
Especially in the newborn with blistering, traumatic, metabolic, hematologic, infectious, drug and autoimmune genesis should be excluded first [15]. Subsequently, a diagnostic algorithm is applied:
- (Family) history and clinic
- Determination of microbial contamination (e.g., smears, PCR, serology).
- Perilesional biopsy for histologic evaluation of possible differential diagnoses.
- Paralesional direct immunofluorescence to determine cleavage level and semiquantitative protein expression from an induced (e.g., rotation of a pencil eraser until erythema develops) and fresh (not older than 12 hours) blister on non-sun-exposed skin.
- Transmission electron microscopy for the determination of the cleavage plane and morphological defects
- Mutation analysis (case-dependent as whole genome, exome, cluster, panel sequencing).
According to the collected findings, the respective EB (sub)type can be determined. The cleavage plane determined by transmission electron microscopy and immunofluorescence defines the main EB type. The clinical phenotype is defined by indicating the relative severity (mild, intermediate, severe) and the pattern of distribution (localized, generalized) – characteristic symptoms such as pseudosyndactyly are also noted if applicable. In addition to specific findings collected in transmission electron microscopy or immunofluorescence mapping, the specific protein and gene affected, mutation type, or specific mutation are cited [9]. The success in molecular characterization of the pathogenetic basis of the different EB types also allows more precise genetic counseling, prognostication and prenatal diagnosis, as well as being a prerequisite for innovative targeted therapeutic interventions.
Therapy principles
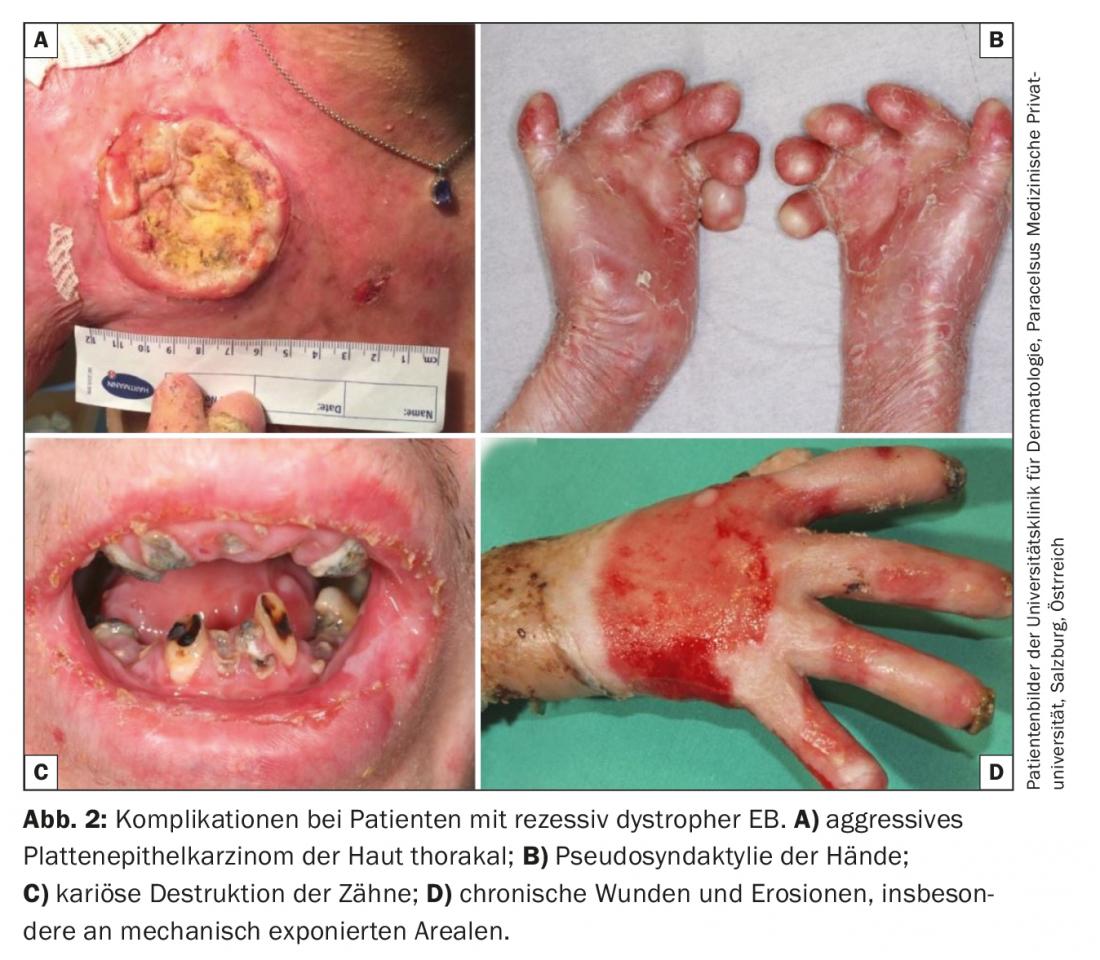
In the absence of curative treatment options in clinical practice, the focus of therapy for all EB forms is on the avoidance of provoking factors and the optimization of symptomatic therapeutic approaches . These include adequate local therapy appropriate to the stage of the wound, improvement or maintenance of the skin barrier through optimized skin care, relief of pain and itching, prevention and treatment of infections (antiseptic and intermittent antibiotic therapy), maintenance of adequate caloric and nutrient intake, and finally, prevention and treatment of complications such as anemia, osteoporosis, and aggressive squamous cell carcinoma. (Fig. 2) [16,17].
The perspectives of molecular, potentially also curative therapy options in EB are in principle encouraging, even if their broad, safe, (sustainably) efficient and practicable implementation in clinical routine for EB patients is currently still difficult to assess. The spectrum of methods is complex:
Gene therapy: In 2006, gene therapy using transplantation of LAMB3 gene-corrected keratinocytes was performed for the first time in a patient with autosomal recessive JEB (mutation in the LAMB3 gene) [18]. In this “gene replacement” therapy, cultured skin cells were transfected with a vector containing a functional cDNA copy of the causative mutant gene and transplanted onto wound areas as epithelial skin equivalents. The dermo-epidermal junctional zone has shown sustained structural and functional stability during the 14-year follow-up period to date, with no evidence of blistering, inflammation, or immune response to the therapeutically introduced neoantigen. In a recent study, autologous keratinocytes were cultured according to the same principle, epidermal stem cells were isolated and selectively gene-corrected, expanded and then transplanted onto large wound areas of a 7-year-old boy with JEB (mutation in the laminin 332 gene). Thus, re-epithelialization was achieved to the extent of 80% of the body surface. The clinical response correlated with the persistent expression of laminin 332 to date [19]. Efficiency limiting factors are a (too) low number of epidermal stem cells in the primary cultures of EB patients to ensure permanent correction, which are depleting among others due to chronic wounds and with advancing age, as well as safety aspects (with respect to viral vectors and genotoxicity or induced insertional mutagenesis). Technological developments are being evaluated in current studies and should improve the risk-benefit profile of this method, which is particularly suitable for circumscribed chronic wounds that are highly symptomatic or at risk of long-term complications [20].
“Gene silencing” for autosomal dominant forms of EB (i.e., silencing the mutant allele with “small interfering RNAs”), as well as application-friendly, topical, gene therapy approaches are also in development. In the case of the latter, for example extracellular vesicles are used, which are isolated from allogeneic mesenchymal stem cells and can transport both the missing type 7 collagen protein and COL7A1 mRNA to target cells [21, 22].
Genome editing technologies using properties of programmable nucleases (e.g. CRISPR/Cas9, TALEN, zinc finger nucleases) are based on the modification/correction of mutated gene sequences. In this process, a double-strand break is specifically induced on the DNA, the mutated sequences are deleted and corrective foreign sequences are inserted, or individual bases are corrected by cellular replication mechanisms. However, safety concerns, particularly regarding insufficient precision and potential off-target binding sites, do not currently justify (in vivo) use in humans [23,24].
A remarkable clinical phenomenon that can occur in all EB major forms is the (spontaneous) occurrence of healthy skin areas without blistering as a result of corrective genetic events, which is called revertant mosaicism or “natural gene therapy”. Attempts to transplant revertant keratinocytes obtained in phase I clinical trials using punch biopsies to affected EB wounds after in vitro expansion have so far fallen short of expectations, primarily due to progressive loss of revertant cells [25,26].
Cell therapy: First results of bone marrow stem cell transplantation showed donor cells in the skin (presumably reprogrammed pluripotent bone marrow stem cells) and good response partly over several years in some patients with recessive-dystrophic EB despite the lack of evidence of a restituted type 7 collagen concentration. This success, the underlying mechanism of which remains unclear, was diminished by significantly increased periprocedural mortality. Revised conditioning therapies and transplantation protocols should improve tolerability [27,28].
Substrates of alternative cell therapies include intradermally or intravenously applied allogeneic mesenchymal stromal cells and adipogenic mesenchymal stem cells. Improved wound healing and reduction of inflammatory signs on the skin could thus be observed, at least temporarily, but this was probably primarily due to the induction of favorable immunomodulatory processes [29]. A clinical effect has also already been demonstrated in initial studies with intradermally injected wild-type or gene-corrected autologous fibroblasts, which also produce type 7 collagen in addition to keratinocytes [30,31].
Cells similar to pluripotent embryonic stem cells can also be derived from somatic cells (e.g. fibroblasts or keratinocytes) by transfection of three to four embryonic transcription factors. The use of these so-called induced pluripotent stem cells (iPSCs), which can differentiate again into various cell types (e.g. keratinocytes or fibroblasts), is also being investigated in EB in preclinical studies. The use of revertant cells/keratinocytes for the production of iPSCs has great potential, since gene correction and the associated risks can be dispensed with [32,33].
Protein therapies: Substitution of the missing or defectively produced protein is also attempted to correct the defective dermo-epidermal junction zone. While studies using recombinant type 7 collagen are still in the preclinical phase, the topical application of a gel containing a modified type 7 collagen-expressing herpes simplex type 1 virus, for example, is already being tested in clinical trials [34,35].
RNA-based therapies/”small molecules”: Nonsense mutations, which cause a stop codon through a DNA point mutation and thus abort translation, are responsible for about 10% of all human genetic diseases. Drugs such as aminoglycosides (e.g., gentamycin) or the immunomodulator amlexanox can lead to “read-through” of the stop codon (e.g., the COL7A1 mutation) by binding to ribosomes in the presence of this mutation, allowing the production of functional proteins [36].
Modifications at the RNA level are also achieved using “antisense oligonucleotide-mediated exon skipping” (targeted removal of exons containing mutations) and “spliceosome-mediated RNA trans-splicing” (SMaRT) (correction of mutated pre-RNA segments). The latter technology has already been used preclinically to successfully correct a mutation of the plectin gene in EB simplex and a COL7A1 mutation in recessive dystrophic EB, as well as autosomal dominant mutations in the keratin-14 gene of an EB simplex cell line [37,38].
So-called “small molecules” are used as mediators of a “disease-modifying” effect. These include topical calcipotriol, which is thought to strengthen endogenous antimicrobial defenses and improve wound healing by increasing expression of the antimicrobial peptide cathelicidin [39] as well as the topical diacerein, a component of rhubarb root and a potent inhibitor of the proinflammatory IL-1β, which has been shown to reduce blister formation in EBS patients, according to preliminary published data [40]. The oral tyrosine kinase inhibitor rigosertib also shows selective inhibition of squamous cell tumor cells from patients with recessive dystrophic EB in vitro, and its efficacy and safety are being evaluated in an ongoing study. Highly aggressive squamous cell carcinoma as a complication of chronic EB wounds is a leading cause of death, particularly in patients with recessive dystrophic EB [41]. In addition to rigosertib, the anti-PD-1 receptor monoclonal antibody nivolumab is currently undergoing controlled testing for efficacy in locally advanced and metastatic squamous cell carcinoma in the EB cohort (EudraCT 2016-002811-16).
Take-Home Messages
- Epidermolysis Bullosa (EB) is a geno- and phenotypically heterogeneous disease. Severe courses develop into a multisystem disease with pronounced morbidity and mortality. The main causes of death are infections, dystrophy, organ failure, and squamous cell carcinoma. The latter occur early and multiply in chronic wounds and show an aggressive course.
- Diagnosis is made by correlation of clinic, histology, immunofluorescence and molecular analysis. Despite innovative, partly causal approaches of molecular therapy strategies, cure is an indirectly still vague future prospect. Immunomodulation, meanwhile, is a hopeful treatment strategy.
- Secondary epigenetic and biochemical components as well as environmental factors that can induce e.g. chronic and systemic inflammatory cascades have high pathogenetic relevance. As with other rare diseases, certain characteristics make it difficult to conduct clinical trials and thus generate high-quality evidence.
Literature:
- Fine JD: Inherited epidermolysis bullosa. Orphanet J Rare Dis 2010; 5: 12.
- Fine JD, et al: Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol 2009; 60(2): 203-211.
- Fine JD, et al: The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 2008; 58(6): 931-950.
- Bruckner-Tuderman L, et al: Progress in epidermolysis bullosa research: summary of DEBRA International Research Conference 2012. J Invest Dermatol 2013; 133(9): 2121-2126.
- Uitto J, Richard G: Progress in epidermolysis bullosa: genetic classification and clinical implications. Am J Med Genet C Semin Med Genet 2004; 131C(1): 61-74.
- Kuttner V, et al: Global remodelling of cellular microenvironment due to loss of collagen VII. Mol Syst Biol 2013; 9: 657.
- Odorisio T, et al: Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-beta signalling in modifying disease severity. Hum Mol Genet 2014; 23(15): 3907-3922.
- Pulkkinen L, et al: Novel ITGB4 mutations in lethal and nonlethal variants of epidermolysis bullosa with pyloric atresia: missense versus nonsense. Am J Hum Genet 1998; 63(5): 1376-1387.
- Fine JD, et al: Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70(6): 1103-1126.
- He Y, et al: Monoallelic Mutations in the Translation Initiation Codon of KLHL24 Cause Skin Fragility. Am J Hum Genet 2016; 99(6): 1395-1404.
- Lee JYW, et al: Mutations in KLHL24 Add to the Molecular Heterogeneity of Epidermolysis Bullosa Simplex. J Invest Dermatol 2017; 137(6): 1378-1380.
- Lin Z, et al: Stabilizing mutations of KLHL24 ubiquitin ligase cause loss of keratin 14 and human skin fragility. Nat Genet 2016; 48(12): 1508-1516.
- Vahidnezhad H, et al: Recessive mutation in tetraspanin CD151 causes Kindler syndrome-like epidermolysis bullosa with multi-systemic manifestations including nephropathy. Matrix Biol 2018; 66: 22-33.
- Salo AM, et al: A connective tissue disorder caused by mutations of the lysyl hydroxylase 3 gene. Am J Hum Genet 2008; 83(4): 495-503.
- Nischler E, et al: Diagnostic pitfalls in newborns and babies with blisters and erosions. Dermatol Res Pract 2009; 2009: 320403.
- El Hachem M, et al: Multicentre consensus recommendations for skin care in inherited epidermolysis bullosa. Orphanet J Rare Dis 2014; 9: 76.
- Dănescu S, et al: Correlation between disease severity and quality of life in patients with epidermolysis bullosa. J Eur Acad Dermatol Venereol 2018. doi: 10.1111/jdv.15371. [Epub ahead of print]
- Mavilio F, et al: Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med 2006; 12(12): 1397-1402.
- Hirsch T, et al: Regeneration of the entire human epidermis using transgenic stem cells. Nature, 2017; 551(7680): 327-332.
- Siprashvili Z, et al: Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016; 316(17): 1808-1817.
- Rosa J, et al: Current Non-viral siRNA Delivery Systems as a Promising Treatment of Skin Diseases. Curr Pharm Des 2018; 24(23): 2644-2663.
- McBride JD, et al: Dual mechanism of type VII collagen transfer by bone marrow mesenchymal stem cell extracellular vesicles to recessive dystrophic epidermolysis bullosa fibroblasts. Biochimie 2018; 155: 50-58.
- Hainzl S, et al: COL7A1 editing via CRISPR/Cas9 in Recessive Dystrophic Epidermolysis Bullosa. Mol Ther 2017; 25(11): 2573-2584.
- March OP, Reichelt J, Koller U: Gene editing for skin diseases: designer nucleases as tools for gene therapy of skin fragility disorders. Exp Physiol 2018; 103(4): 449-455.
- Gostynski A, Pasmooij AM, Jonkman MF: Successful therapeutic transplantation of revertant skin in epidermolysis bullosa. J Am Acad Dermatol 2014; 70(1): 98-101.
- van den Akker PC, et al: A “late-but-fitter revertant cell” explains the high frequency of revertant mosaicism in epidermolysis bullosa. PLoS One 2018; 13(2): p. e0192994.
- Ebens CL, et al: Bone marrow transplant with post-transplant cyclophosphamide for recessive dystrophic epidermolysis bullosa expands the related donor pool and permits tolerance of non-hematopoietic cellular grafts. Br J Dermatol 2019. doi: 10.1111/bjd.17858. [Epub ahead of print]
- Vanden Oever M, et al: Inside out: regenerative medicine for recessive dystrophic epidermolysis bullosa. Pediatr Res 2018; 83(1-2): 318-324.
- Ganier C, et al: Intradermal Injection of Bone Marrow Mesenchymal Stromal Cells Corrects Recessive Dystrophic Epidermolysis Bullosa in a Xenograft Model. J Invest Dermatol 2018; 138(11): 2483-2486.
- Petrof G, et al: Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomized, vehicle-controlled trial. Br J Dermatol 2013; 169(5): 1025-1033.
- Venugopal SS, et al: A phase II randomized vehicle-controlled trial of intradermal allogeneic fibroblasts for recessive dystrophic epidermolysis bullosa. J Am Acad Dermatol 2013; 69(6): 898-908.e7. doi: 10.1016/j.jaad.2013.08.014
- Itoh M, Kiuru M, Cairo MS, Christiano AM.: Generation of keratinocytes from normal and recessive dystrophic epidermolysis bullosa-induced pluripotent stem cells. Proc Natl Acad Sci U S A 2011; 108(21): 8797-8802.
- Nakayama C, et al: The development of induced pluripotent stem cell-derived mesenchymal stem/stromal cells from normal human and RDEB epidermal keratinocytes. J Dermatol Sci 2018; 91(3): 301-310.
- South AP, Uitto J: Type VII Collagen Replacement Therapy in Recessive Dystrophic Epidermolysis Bullosa-How Much, How Often? J Invest Dermatol 2016; 136(6): 1079-1081.
- NIH: Topical Bercolagene Telserpavec (KB103) Gene Therapy to Restore Functional Collagen VII for the Treatment of Dystrophic Epidermolysis Bullosa (GEM-1), https://clinicaltrials.gov/ct2/show/NCT03536143, last accessed Mar 25, 2019.
- Lincoln V, et al: Gentamicin induces LAMB3 nonsense mutation readthrough and restores functional laminin 332 in junctional epidermolysis bullosa. Proc Natl Acad Sci U S A, 2018; 115(28): E6536-E6545.
- Peking P, et al: A Gene Gun-mediated Nonviral RNA trans-splicing Strategy for Col7a1 Repair. Mol Ther Nucleic Acids 2016. 5:e287. doi: 10.1038/mtna.2016.3.
- Turczynski S, et al: Targeted Exon Skipping Restores Type VII Collagen Expression and Anchoring Fibril Formation in an In Vivo RDEB Model. J Invest Dermatol 2016; 136(12): 2387-2395.
- Guttmann-Gruber C, et al: Low-dose calcipotriol can elicit wound closure, anti-microbial, and anti-neoplastic effects in epidermolysis bullosa keratinocytes. Sci Rep 2018; 8(1): 13430.
- Wally V, et al: Diacerein orphan drug development for epidermolysis bullosa simplex: A phase 2/3 randomized, placebo-controlled, double-blind clinical trial. J Am Acad Dermatol 2018; 78(5): 892-901.e7. doi: 10.1016/j.jaad.2018.01.019.
- Atanasova VS, et al: Identification of rigosertib for the treatment of recessive dystrophic epidermolysis bullosa-associated squamous cell carcinoma. Clin Cancer Res, 2019. doi: 10.1158/1078–0432.CCR-18–2661
DERMATOLOGIE PRAXIS 2019; 29(2): 16-20









