Lysosomal storage diseases are a heterogeneous group of genetic disorders that originate from dysfunction of lysosomal metabolic processes. Niemann-Pick disease is nowadays also referred to as ASMD (“Acid Sphingomeyelinase Deficiency”). Type A and B are classified as sphingolipidoses, while type C belongs to lipid storage diseases. For type A and B, the first enzyme replacement therapy was approved in the EU last year.
Niemann-Pick disease is a genetic lysosomal storage disorder named after the German pediatrician Albert Niemann (1880-1921) and the German pathologist Ludwig Pick (1868-1944). Ludwig Pick succeeded in distinguishing Niemann-Pick disease as an independent metabolic disorder from Gaucher disease [1]. The storage substance sphingomyelin was detected by the biochemist Klenk in 1934. A classification into different subgroups was initiated by Crocker in 1961 [2–4]. The most common forms of manifestation are type A-C (box).
| The most common subtypes Niemann-Pick syndrome is an autosomal recessive inherited disease. Type A and B are due to deficient activity of a lysosomal enzyme encoded by the SMPD1 gene. The genetic defect causes sphingomyelin to be unable to be broken down and to accumulate in the cells of various organs. Type C is a cholesterol metabolism disorder in which mutations are detectable in the NPC-1 gene (18q11) or the NPC-2 gene (14q24.3). |
| to [11] |
Clinical manifestations
Type A is a severe neurodegenerative disease of infancy that usually leads to death within the first three years of life. The main symptoms are hepatosplenomegaly and psychomotor decline. The development of the affected children stagnates. Skills learned in previous years of life are lost over time. Frequently, failure to thrive, vomiting, hearing loss, tetraspasticity, and myoclonic seizures can be observed before completion of the first six months of life.
Type B is characterized by a later onset of disease and a milder manifestation than type A [5]. Most patients reach adulthood and no cerebral symptoms are observed. In contrast, hepatosplenomegaly with progressive hypersplenism and stable hepatic dysfunction, as well as a gradual deterioration of pulmonary function, accompanied by osteopenia and an atherogenic lipid profile, are characteristic [6]. A proatherogenic lipid profile is seen early in the disease course, and some patients develop coronary artery disease.
Type C is associated with impaired cholesterol transport from lysosomes, resulting in increased storage of cholesterol, gycosphingolipids, and gangliosides in lysosomes of various body cells [7,8]. Compared to type A/B, the clinical presentation is very heterogeneous. It is a chronic neurovisceral disease that is more slowly progressive compared to type A. It can be divided into early infantile, late infantile, juvenile, and adult forms [7]. Clinically, affected individuals present with various neurological and psychiatric abnormalities, and sometimes visceral symptoms such as hepatosplenomegaly [8]. Typical neurological manifestations in type C include cognitive impairment, epileptic seizures, behavioral abnormalities, depression and psychosis, vertical gaze palsy, speech and swallowing disorders, and dystonia [9].
| Type A/B: new enzyme replacement therapy: olipudase alfa For pediatric and adult patients with Niemann-Pick disease or ASMD (“acid sphingomeyelinase deficiency”) type A/B or type B without central nervous involvement, the enzyme replacement therapy olipudase alfa was approved by the EMA in 2022. Olipudase alfa is designed to replace absent or defective ASM to allow sphingomyelin degradation. The approval decision is based on data from the ASCEND and ASCEND-Peds clinical trials, which showed clinically relevant improvements in lung function and reductions in spleen and liver volume with olipudase alfa therapy. The incidence of adverse events in patients receiving olipudase alfa was comparable to the placebo group. Olipudase alfa is infused every two weeks in the maintenance phase. In the ASCEND study, 36 adult patients with ASMD type A/B or type B were randomized to olipudase alfa or placebo. After 52 weeks, the treatment arm showed improvement in lung function and reduced spleen volume. In the single-arm ASCEND-PEDS study, 20 pediatric patients with ASMD type A/B or type B were treated with olipudase alfa for 64 weeks. Again, the key endpoints were met at week 52. |
| to [12] |
Diagnostics and therapy
Reduced or absent acid sphingomyelinase activity can be detected in leukocyte and fibroblast cultures, and the cause of Niemann-Pick type A and B disease is [10]. These enzymatic and molecular genetic tests can be performed prenatally if a family history is known [1]. To establish the diagnosis of Niemann-Pick type C disease, elaborate investigations of cholesterol metabolism must be performed [10]. The underlying genetic defects are currently not treatable (as of 2022). For Niemann-Pick disease types A and B, enzyme replacement therapy with olipudase alfa is available in the EU (box). Type C is treated symptomatically with miglustat.
Literature:
- “Study of the liver and spleen by elastography in Niemann-Pick type B disease patients,” Gözde Aksu, Inaugural Dissertation, 2020, https://openscience.ub.uni-mainz.de, (last accessed Oct. 12, 2023).
- Crocker AC, Mays VB: Sphingomyelin synthesis in Niemann-Pick disease. Am J Clin Nutr 1961; 9: 63-67.
- Crocker AC: The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. J Neurochem 1961; 7: 69-80.
- E. K. On the nature of phosphatides of the spleen in Niemann-Picksen disease. Hoppe Seyler’s Journal of Physiological Chemistry. 1934.
- Tran C, et al: Pulmonary involvement in adult patients with inborn errors of metabolism. Karger Compass Pneumol 2018; 6: 6-17.
- Orphanet, www.orpha.net,(last accessed Oct. 12 , 2023).
- Di Lazzaro V, et al: Niemann-Pick type C: focus on the adolescent/adult onset form. Int J Neurosci 2016; 126(11): 963-971.
- Hammerschmidt TG, et al: Molecular and biochemical biomarkers for diagnosis and therapy monitorization of Niemann-Pick type C patients. Int J Dev Neurosci 2017; 66: 18-23.
- Bonnot O, et al: Psychiatric and neurological symptoms in patients with Niemann-Pick disease type C (NP-C): Findings from the International NPC Registry. World J Biol Psychiatry 2017: 1-10.
- “Niemann-Pick’s disease,” https://flexikon.doccheck.com,(last accessed Oct. 12, 2023).
- Desnick JP, et al: Identification and characterization of eight novel SMPD1 mutations causing types A and B Niemann Pick disease. Mol Med 2010; 16: 316-321.
- “Xenpozyme® (olipudase alfa) approved by European Commission as first and only treatment for ASMD”, 06/28/2022.
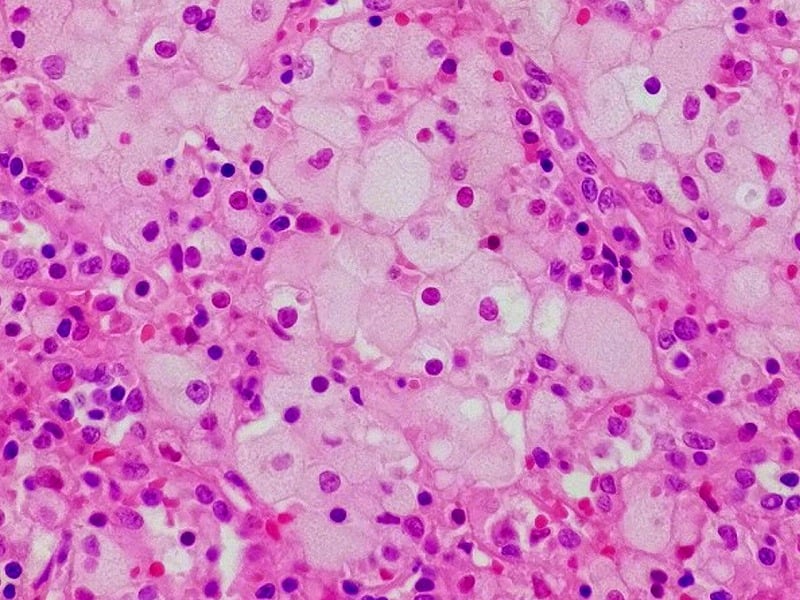
| Cover picture: Niemann pick cell in spleen. ©W.CC, Wikimedia |
GP PRACTICE 2023; 18(10): 48