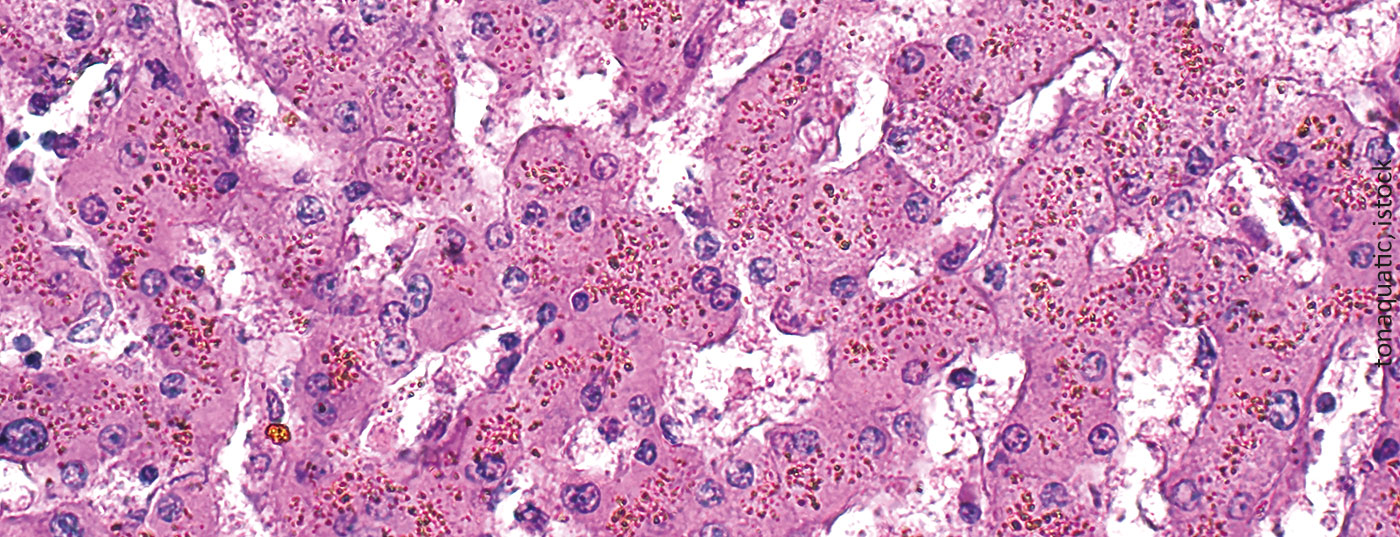
Iron overload is an unavoidable and potentially life-threatening consequence of multiple red cell concentrate transfusions. Because the clinical manifestations are nonspecific and usually develop slowly, this complication is often overlooked.
Iron overload is an unavoidable and potentially life-threatening consequence of multiple red cell concentrate transfusions. Because the clinical manifestations are nonspecific and usually develop slowly, this complication is often overlooked.
Generally, a distinction between primary and secondary iron overload is no longer made today because such a definition depends, among other things, on the sensitivity of the examination methods. “Siderosis” usually refers to iron deposition without tissue damage, as occurs with localized, hemorrhage-related iron deposition (e.g., pulmonary siderosis).
Emergence of iron overload
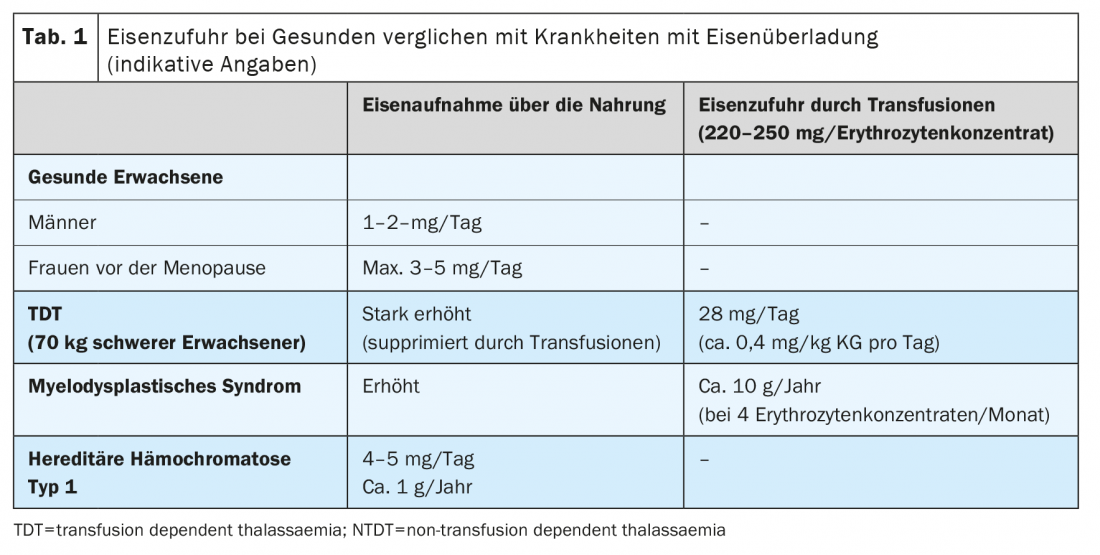
A typical Western diet contains about 6 mg iron/1000 kcal, of which only 1-2 mg/day (about 10%) is normally absorbed and a maximum of 3-5 mg/day is absorbed when intestinal iron absorption is upregulated. Normal storage iron is 500-1000 mg in men and 300-400 mg in pre-menopausal women, and most of it is found in the liver. The organism has no active physiological mechanisms to excrete excess iron. Healthy adults excrete about 1 mg of iron/day through the skin and gastrointestinal cells, and pre-menopausal women lose an additional 0.5-1 mg/day on average through menstruation.
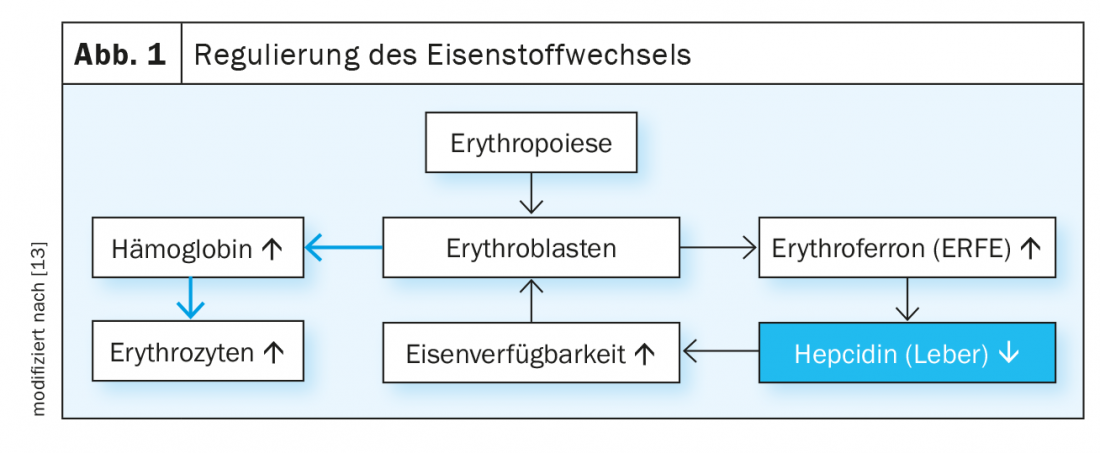
Plasmatic iron levels are regulated by the hepcidin/ferroportin system (Fig. 1). The peptide hormone hepcidin induces the degradation of the iron export protein ferroportin. Ferroportin is expressed primarily in duodenal mucosal cells, liver cells, and macrophages and mediates the regulation of dietary iron absorption, hepatic iron release on demand, and iron recycling in macrophages. With sufficient iron availability, hepatic synthesis of hepcidin increases, thereby blocking further gastrointestinal iron absorption. Chronic anemia and ineffective erythropoiesis, on the other hand, lead to inhibition of hepcidin synthesis in the liver, resulting in increased iron absorption in the duodenum. The hormone erythroferron, which is produced under the influence of erythropoietin in erythroblasts, also suppresses the production of hepcidin and thus stimulates iron absorption and mobilization from stores in situations of erythropoietic stress.
Excessive iron storage is due to two main mechanisms: The iatrogenic supply of iron by transfusions of red cell concentrates and the increased intake of iron from food. Each red cell concentrate contains 220-250 mg of iron. In adult patients, relevant iron deposition occurs after 15-20 transfusions, in young children after more than 10 administrations of red blood cell concentrates. Increased gastrointestinal absorption plays a central role in all situations of increased or ineffective erythropoiesis. Here, erythroblasts perish by cell death in the bone marrow before they can mature into erythrocytes. This results in hyperstimulation and hyperplasia of the erythropoiesis, which varies depending on the underlying disease. The more pronounced the impairment of erythropoiesis, the greater the upregulation of dietary iron absorption (Table 1).
In hereditary hemochromatosis, a genetic disorder of iron metabolism without anemia, iron deposition results from dysregulation of the hepcidin-ferroportin axis, most commonly from decreased hepcidin production (as in hereditary hemochromatosis type 1). Less commonly, dysfunction of other iron-regulating molecules is present.
Depending on the disease, the time course and extent of iron accumulation as well as the distribution of iron in the organs differ. This is also influenced by other factors such as chronic infections (e.g. chronic hepatitis) and metabolic diseases (e.g. steatohepatitis).
Effects of excess iron on cells
Iron-associated toxicity is observed primarily in those tissues that store iron in high concentrations. These are in particular the liver, the endocrine system and the myocardium. Iron from transfusions is deposited first in macrophages of the liver, spleen, and bone marrow, then in hepatocytes, and only later in endocrine organs (especially the pancreas) and cardiac muscle.
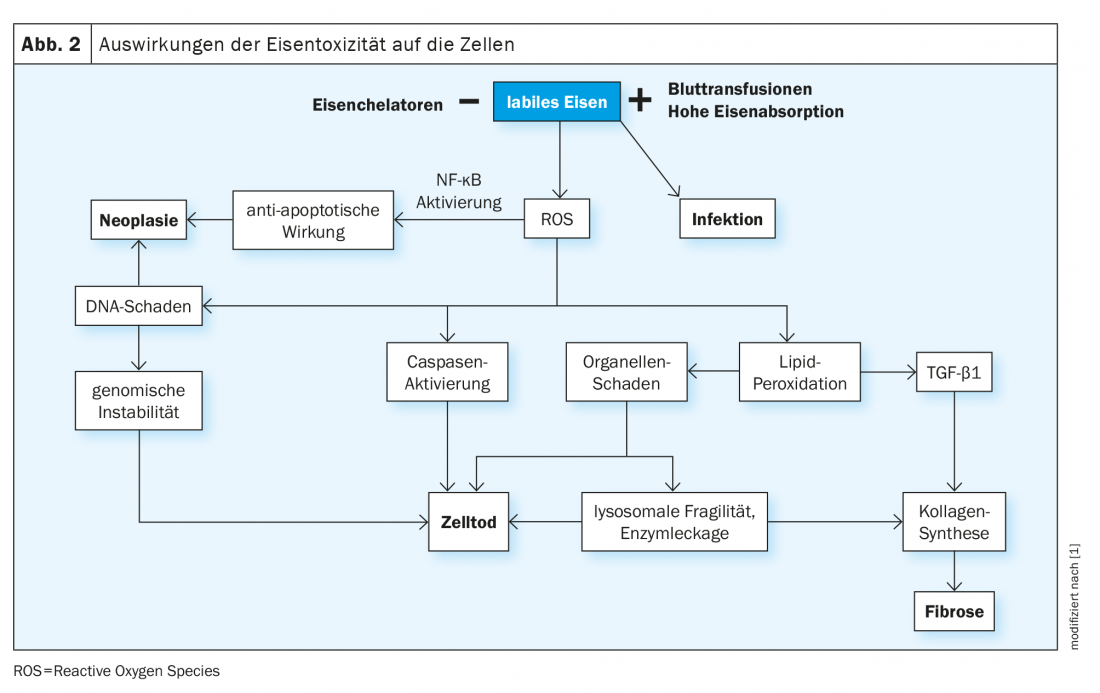
Circulating iron is bound by the protein transferrin and then delivered to cells by the binding of transferrin to the appropriate receptors (TfR1, expressed on all cells, and TfR2, present on hepatocytes). The normal percentage of transferrin bound to iron is 16-45% and is measured as transferrin saturation. In the presence of excess iron, the binding capacity of transferrin is exceeded and at levels above 70-75%, circulating unbound iron molecules (non-transferrin bound iron , NTBI) appear and are rapidly taken up into cells via unregulated pathways (e.g., via calcium channels into cells of the myocardium). Continued uptake of NTBI increases both physiological intracellular iron stores (ferritin) and the occurrence of labile forms of iron. The latter catalyze biochemical reactions such as the Fenton reaction, producing reactive oxygen radicals (ROS). The consequences are peroxidation of intracellular molecules, especially lipids, with damage to organelles, cell death, stimulation of fibrogenesis, and finally organ dysfunction. Furthermore, DNA is damaged, which can lead to genome instability and a propensity for mutagenesis (Fig. 2) .
Iron deposition is tolerated differently by different tissues. For example, the liver can store much more iron without harmful consequence than the myocardium. Another critical factor in the development of organ damage is the duration of cell exposure to NTBI. This can now persist for several decades in congenital transfusion-dependent diseases, such as thalassemia major, and is associated with an increased risk of malignancy.
An underestimated effect of iron overload is the favoring of bacterial growth and the corresponding risk for infections. The rapid availability of free iron to microorganisms, on the one hand, and the effects on macrophage and leukocyte function, on the other, explain the increased susceptibility of patients with transfusion-dependent thalassemia to infection. Recent observations also show that excess iron causes oxidative stress on the endothelium of all vessels and decreased cholesterol export by macrophages in the vessel wall, which may lead to increased plaque formation.
Patients at risk for iron overload
Clinical situations associated with iron overload include congenital and acquired diseases with ineffective erythropoiesis (e.g., thalassemias and myelodysplastic syndrome), chronic hemolytic disease, sickle cell disease (SCD) requiring transfusion, and genetic disorders of iron metabolism without anemia (hereditary hemochromatosis) (Tab. 2). In addition, iron excess is of prognostic significance in patients after allogeneic stem cell transplantation.
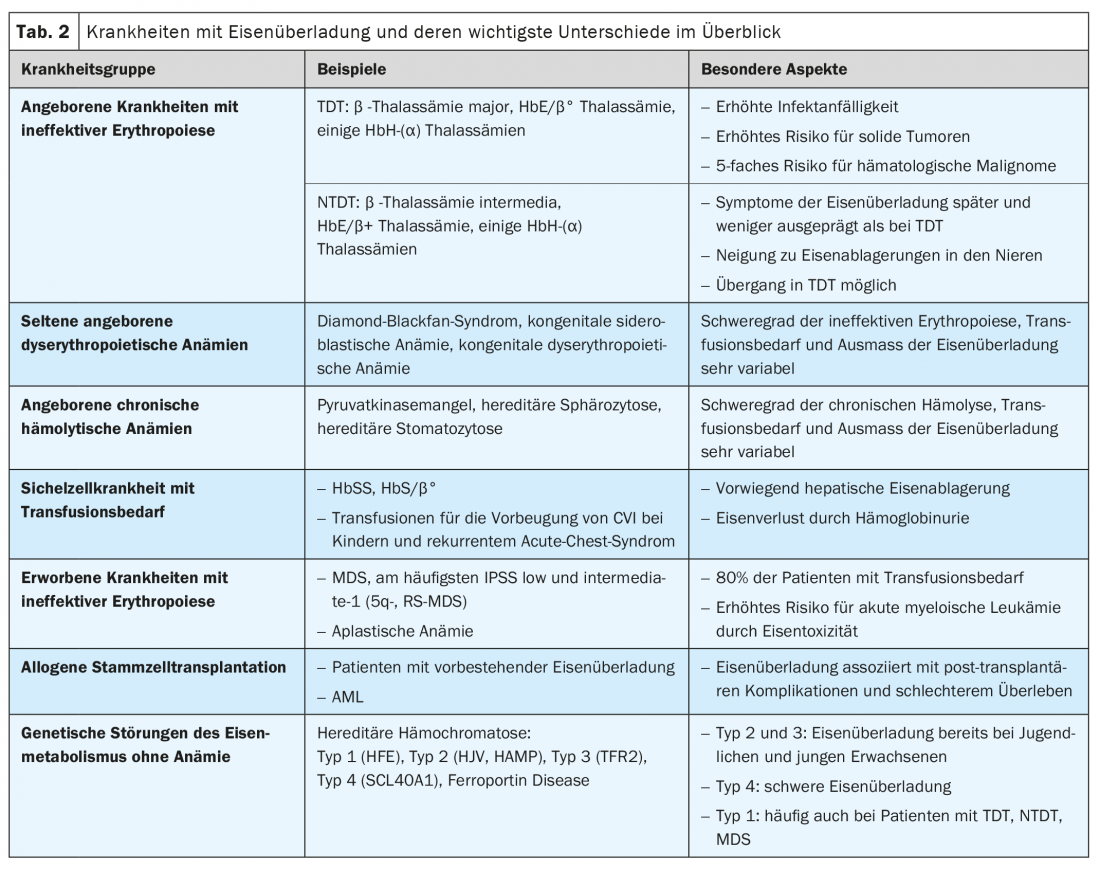
Transfusion-dependent (TDT) and non-transfusion-dependent thalassemia (NTDT).
Thalassemia syndromes are among the most common genetic diseases worldwide and are caused by gene defects on chromosome 11(β-thalassemia) or 16(α-thalassemia) that result in decreased or absent synthesis of the corresponding globin chains. The enormous number of genetic variants and their combinations described so far, as well as the type of inheritance, explain the high clinical variability. From a clinical perspective, transfusion-dependent (TDT) and non-transfusion-dependent (NTDT) thalassemias are distinguished. This subdivision corresponds approximately to that in Thalassämia major and intermedia.
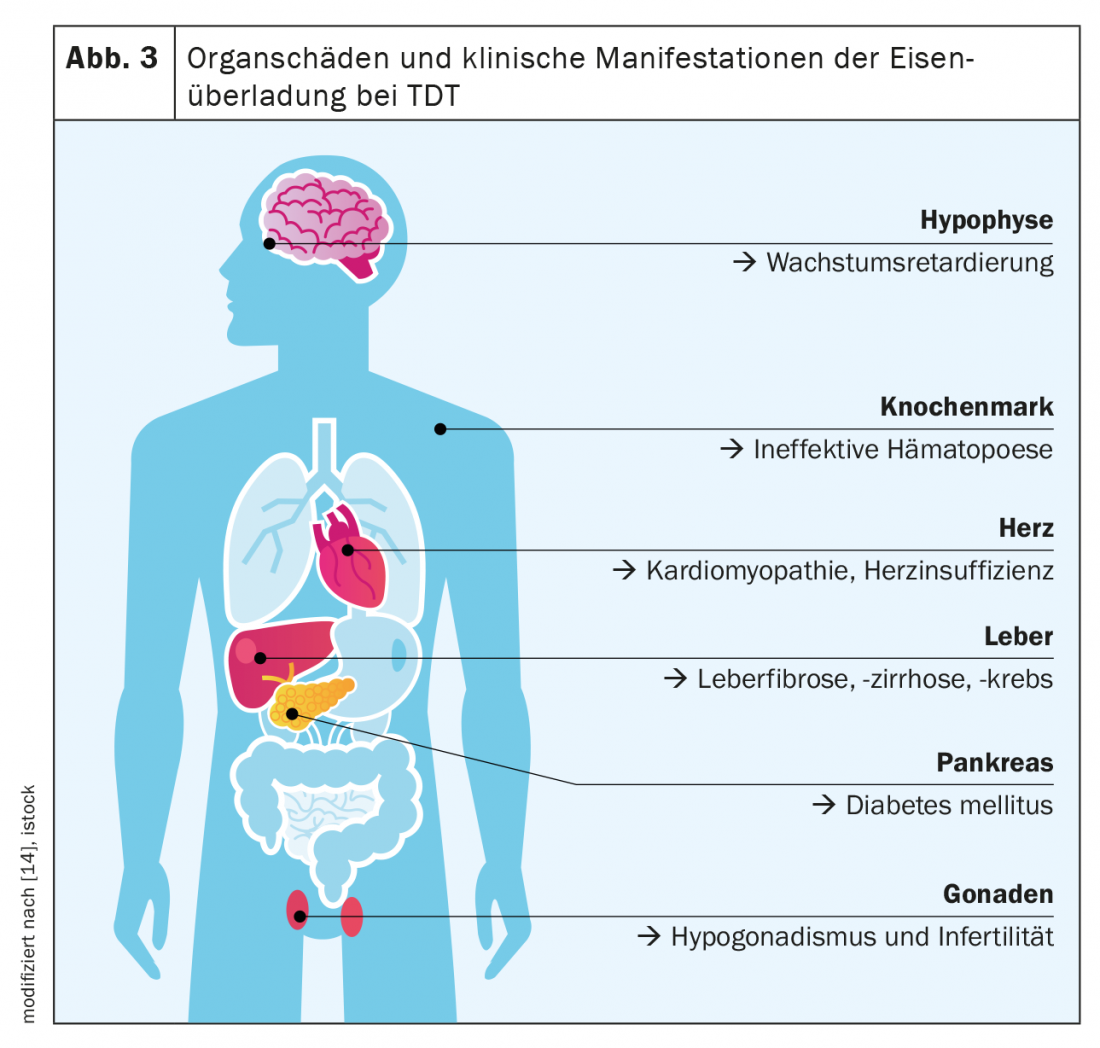
The β-thalassemia major is considered the paradigm of TDT and iron overload diseases. The main feature of TDT is severely impaired to absent erythropoiesis, which results from decreased hemoglobin synthesis and precipitation of excess globin chains(α or β). There is severe anemia with, on the one hand, a lifelong need for transfusions from the first year of life and, on the other hand, maximally stimulated intestinal iron absorption. Thus, massive daily dietary iron intake and iatrogenic iron intake, which alone is about 0.4 mg/kg bw per day, add up. Classic symptoms of iron overload in TDT are hepatopathy (fibrosis, cirrhosis, hepatocellular carcinoma), cardiopathy (especially rhythmogenic and dilated cardiopathy), endocrinopathies (diabetes mellitus, hypothyroidism, hypogonadism), and osteoporosis ( Fig. 3) .
With the introduction of transfusion therapy in the 1970s, the life expectancy of patients with TDT was extended to puberty, with cardiac and endocrine failure being the most common causes of death. Thanks to the possibility of iron chelation, patients with TDT now have a life expectancy of 40-50 years or longer and have less severe forms of cardiopathies and endocrinopathies. However, there is an increased risk of malignancies, particularly gastrointestinal carcinomas, and a more than 5-fold increased risk of hematologic neoplasms as a long-term consequence of iron toxicity.
In NTDT, increased iron absorption (approximately 0.01 mg/kg bw per day) is the consequence of greatly increased erythropoiesis and represents the main mechanism of iron deposition. Because iron accumulation in NTDT develops slowly, clinical manifestations do not occur until later childhood or even early adulthood. Symptoms of iron overload are generally less pronounced, cardiac involvement less common, and the risk of neoplasia lower than in TDT. However, patients with NTDT have a tendency to accumulate iron in the kidneys, which can lead to interstitial and glomerular dysfunction. Importantly, NTDT can transition to TDT over the course of a lifetime.
Rare congenital anemias with ineffective erythropoiesis and congenital chronic hemolytic anemias.
Also, in congenital anemias, such as Diamond-Blackfan syndrome, the severity of ineffective erythropoiesis and anemia, as well as transfusion requirements, determine the extent of iron overload. Specific data are scarce for these rare diseases. Depending on transfusion needs, which may be relevant even in young children, risks and clinical manifestations of iron overload resemble those of TDT and NTDT.
Congenital erythrocytic enzymopathies such as pyruvate kinase deficiency, membranopathies (e.g., hereditary spherocytosis), and disorders of heme synthesis (e.g., porphyrias) result in more or less marked chronic hemolysis and consequent hyperplasia of the erythropoiesis. Affected individuals may experience iron overload even without transfusions (up to 47% of patients with pyruvate kinase deficiency), with corresponding symptoms typically occurring in adulthood and rarely in childhood. In hereditary spherocytosis, which is the most common cause of chronic hemolysis with an estimated prevalence of 1:2000 to 1:2500, only a proportion of patients require transfusions sporadically or regularly.
Sickle cell disease (SCD) with transfusion requirement
Regular transfusions are indicated in patients with SCD only in specific situations, including prevention of cerebrovascular insults in children and recurrent acute chest syndrome or severe vaso-occlusive crises. Manual or automated exchange transfusions are often performed in these cases, which are not associated with increased transfusion-related iron intake.
Patients with SCD frequently lose iron through hemoglobinuria (in amounts equivalent to up to 10 red cell concentrates annually) and have low plasma levels of NTBI. Therefore, iron overload develops in only a relatively small proportion of those affected, and symptoms – especially hepatopathy and liver fibrosis – classically appear later in life. Although endocrine organ or cardiac damage is uncommon in patients with SCD, the clinical manifestations of iron overload and SCD itself are often difficult to distinguish, and therefore iron-related organ damage is sometimes underestimated. For example, studies show that up to 11% of pa-tients with SCD die as a result of iron overload. Due to increasingly longer life expectancy, long-term iron-associated complications are also expected to increase in SCD, as in TDT and NTDT.
Acquired chronic anemia due to bone marrow failure
Myelodysplastic syndrome (MDS) comprises a group of acquired diseases characterized by bone marrow failure of varying degrees and the potential of developing acute myeloid leukemia. MDS is one of the most common hematologic neoplasms (incidence of approximately 4/100,000 in Europe) and mostly affects elderly patients (median age at diagnosis 71 years).
In low-risk MDS (IPSS low- and intermediate-1), cytopenias are prominent, with anemia due to ineffective erythropoiesis occurring in approximately 80% of cases. As most patients with MDS become transfusion dependent, they often develop iron overload. Here, the most important differences from TDT and NTDT are the much older age of the patients at diagnosis and the high genome stability of the dysplastic clones in the bone marrow. As with TDT, cardiac complications are common in regularly transfused MDS patients (82.4% versus 67.1% in nontransfused patients). A particular significance of iron toxicity in MDS is the promotion of progression to acute leukemia and the negative impact on survival after allogeneic stem cell transplantation.
Allogeneic stem cell transplantation
Allogeneic stem cell transplantation is a potentially curative treatment for patients with TDT, SCD, and MDS, as well as other diseases with iron deposition. The impact of iron overload on the success of allogeneic stem cell transplantation, particularly on therapy-associated mortality, was first described in thalassemias and is now increasingly recognized in other disease conditions.
Even the toxicity of chemotherapy can be attributed in part to adverse effects of iron overload, because during conditioning there is a massive mobilization of iron stores from the bone marrow with release of NTBI. Transfusion requirements before and after transplantation influence the peri-transplant course. Data show that 88% of patients with MDS and 97% of patients with acute myeloid leukemia have elevated ferritin levels before transplantation and that iron excess is associated with mucositis, sinusoidal hepatic vessel obstruction, sepsis, and overall lower overall survival.
Genetic disorders of iron metabolism without anemia
Increased intestinal iron absorption is the pathophysiological correlate of the various forms of hereditary hemochromatosis. Iron intake may be increased up to 4 mg daily, 2-4 times higher than in individuals without hereditary hemochromatosis (Table 1) .
In HFE-associated hereditary hemochromatosis (type 1), one of the most common genetic diseases in the European population, iron is deposited very slowly (about 1 g/year) and symptoms typically appear in adulthood (between 40 and 50 years of age in men, often after menopause in women). The most common clinical manifestations are hepatopathy, arthropathies and diabetes mellitus. The timing and severity of iron accumulation differ depending on the underlying mutation. Some rare forms such as juvenile hemochromatosis (type 2) are more aggressive and lead to relevant iron storage as early as puberty.
Because of its frequency, hereditary hemochromatosis, particularly type 1, is not infrequently detected in patients with thalassemia, MDS, or risk for pathologic iron accumulation and thus represents an additional factor in the development of potentially severe iron overload.
Take-Home Messages
- Increased intestinal iron absorption and iatrogenic iron supplementation by transfusions of red blood cell concentrates are the main mechanisms for the development of iron overload.
- Pathological iron accumulation occurs in congenital and acquired hematologic diseases with ineffective erythropoiesis, even in the absence of regular transfusions.
- Iron overload develops more rapidly and to a greater extent in regularly transfused patients than in non-transfusion-dependent diseases; in genetic disorders of iron metabolism (hereditary hemochromatosis), iron is deposited much more slowly in comparison.
- In MDS, cellular iron excess may enhance genome instability in pre-leukemic cell clones, promoting transformation to acute leukemia.
- Iron overload is associated with clinical complications and increased mortality after allogeneic stem cell transplantation.
- Hereditary hemochromatosis, because of its frequency, may be present in patients with other risks for clinically relevant iron overload and may affect the clinical course.
Literature:
- Porter JB, et al: New insights into transfusion-related iron toxicity: implications for the oncologist. Crit Rev Oncology/Hematology 2016; 99: 261-271.
- Camaschella C, Nai A, Silvestri L: Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020; 105: 260-72.
- Porter JB, Garbowski M: The pathophysiology of transfusional iron overload. Hematol. Oncol Clin N Am 2014; 28: 683-701.
- Hahalis G, et al: Global vasomotor dysfunction and accelerated vascular aging in beta-thalassemia major. Atherosclerosis 2008; 198 (2): 448-457.
- Gardenghi S, et al: Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood 2007; 109(11): 5027-5035.
- Vento S, Cainelli F, Cesario F: Infections and thalassaemia. Lancet Infect Dis 2006; 6(4): 226-233.
- Porter J, Garbowski M: Consequences and management of iron overload in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2013; 2013: 447-456.
- Roggero S, et al: Severe iron overload in Blackfan-Diamond anemia: a case-control study. Am J Hematol. 2009; 84: 729-32.
- Zanella S, Garani MC, Borgna-Pignatti C: Malignancies and thalassemia: a review of the literature. Ann N Y Acad Sci 2016; 1368(1): 140-148.
- Gattermann N: Iron overload in myelodysplastic syndromes (MDS). Int J Hematol 2018; 107: 55-63.
- Koreth J, Antin JH: Iron overload in hematologic malignancies and outcome of allogeneic hematopoietic stem cell transplantation. Haematologica 2010; 95: 364-366.
- Pilling LC, et al: Common conditions associated with hereditary haemochromatosis genetic variants: cohort study in UK Biobank. BMJ 2019; 364: k5222.
- Leuenberger N, et al: Hepcidin as a potential biomarker for blood doping. Drug Test Anal 2017; 9(7): 1093-1097.
- Novartis: Living with transfusions. www.leben-mit-transfusionen.de/eisenueberladung/krankheitsbild (last accessed on 10.02.2022)
InFo ONCOLOGY & HEMATOLOGY 2022; 10(1): 12-17.