Amyloidoses are protein misfolding diseases in which extracellular deposition of insoluble protein fibrils with an antiparallel β-sheet configuration usually occurs. The underlying pathomechanisms are diverse. Acquired causes include high levels of the proteins in question in the blood or tissues in an underlying clonal disease or inflammation.
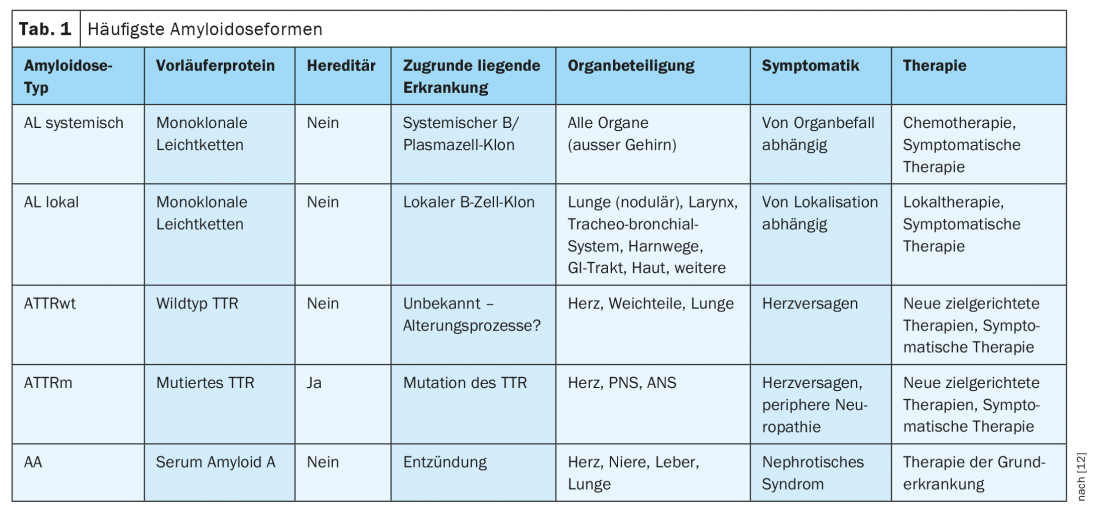
Amyloidoses are protein misfolding diseases in which extracellular deposition of insoluble protein fibrils with an antiparallel β-sheet configuration usually occurs [1]. In humans, 36 amyloidogenic proteins are currently known to be deposited in the form of these aggregates [2]. The underlying pathomechanisms are diverse. Acquired causes include high levels of the proteins in question in the blood or tissues in an underlying clonal disease or inflammation. Hereditary amyloidoses, on the other hand, are mostly due to point mutations with a subsequently altered tertiary structure of the protein in question. The resulting aggregates may be local (identical site of production and deposition) or systemic (different sites of production and deposition) and often show organ tropism characteristic of the corresponding amyloidosis subtype. The naming of the amyloidosis forms is done according to international nomenclature by acronyms, where the capital letter “A” is followed by an abbreviation for the respective precursor protein [2]. The most common amyloidoses and their central characteristics are summarized in Table 1.
The most common amyloidosis in industrialized countries is systemic AL amyloidosis. With about 10 cases per million person-years [3], it is one of the rare diseases. It is usually caused by monoclonal plasma cells in the bone marrow that produce an amyloidogenic light chain. AL amyloid deposits are also found in many cases of local amyloidosis. However, in contrast to systemic AL amyloidosis, AL amyloid is deposited locally or multilocularly and is almost always confined to one organ system. In these cases, clonal plasma or B cells are often present only locally and monoclonal gammopathy is undetectable or only very slightly detectable in blood or urine [4]. Little is known about the genesis of local AL amyloidosis. Presumably, antigen-induced local stimulation of B cells also plays an important role in many cases [5]. In addition, increased B-cell activity in the context of an underlying inflammatory rheumatologic disease may be causative [6,7].
The symptoms and treatment options of each amyloidosis are derived from the underlying pathology and organ involvement. The spectrum ranges from no need for treatment with unrestricted life expectancy and quality of life, to a potentially threatening but easily treatable disease, to the most severe disease with an extremely unfavorable prognosis [8,9]. Unfortunately, due to the rarity of amyloidoses and the often unspecific symptoms, the diagnosis is often made late, at a time when, in the case of systemic amyloidoses, severe and hardly reversible end-organ damage has often already occurred. To confirm the diagnosis (except in ATTR amyloidosis of the heart), histologic evidence of typical birefringent material in Congo red staining is obligatory. Tissue may be obtained from the affected organ or, in the case of systemic amyloidoses, from any other potential site of deposition. In systemic light chain (AL) amyloidosis, adipose tissue aspiration has proven to be a very sensitive method to confirm the diagnosis [10]. For differential diagnosis and exact classification of amyloidosis as well as for the development of an individual treatment strategy, the presentation of the patient in an amyloidosis center is recommended. In particular, the histological subtyping, the clinical constellation of findings, as well as the extent of organ damage and, accordingly, the therapeutic capability are taken into account. Standard for differentiation of the different forms of amyloidosis in Germany is immunohistochemistry, which should be performed by specialized pathologists or institutes [11]. A repeat biopsy is usually not necessary.
Pulmonary amyloidosis
Pulmonary amyloidosis is a very rare diagnosis. However, the exact prevalence is unknown, as it is often asymptomatic and therefore often incidental or even post-mortem findings. From April 2000 to October 2019, we saw a total of 157 patients with local pulmonary amyloidosis at our center [6]. During the same period, we saw 2240 patients with the most common systemic amyloidosis in Germany and Switzerland, AL amyloidosis. Autopsy studies suggest that pulmonary amyloid deposition and consecutive pulmonary tissue changes are detectable in systemic AL amyloidosis in up to 90% [12]. However, the clinical symptoms of affected patients are extremely rarely attributed to lung involvement, even when CT morphologic abnormalities are present [13]. This is due to cardiac involvement, which is clinically leading and easily confirmed by echocardiography in most patients with systemic AL amyloidosis [9]. In contrast, isolated pulmonary symptomatology is more commonly found in local AL amlyoidosis [6]. These also represent the majority of pulmonary amyloidoses diagnosed ante mortem. The life expectancy of patients with local AL amyloidosis is generally not limited compared with the general population. Furthermore, the risk of progression to systemic AL amyloidosis is usually low. However, the clinical course is often characterized by local recurrences or progression.
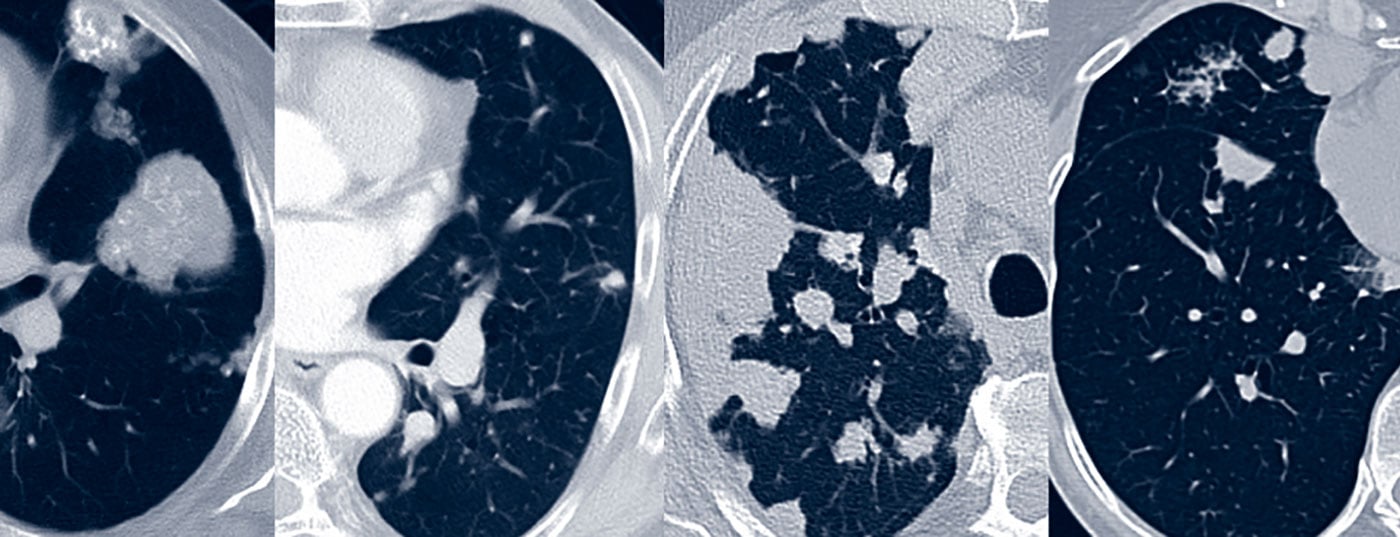
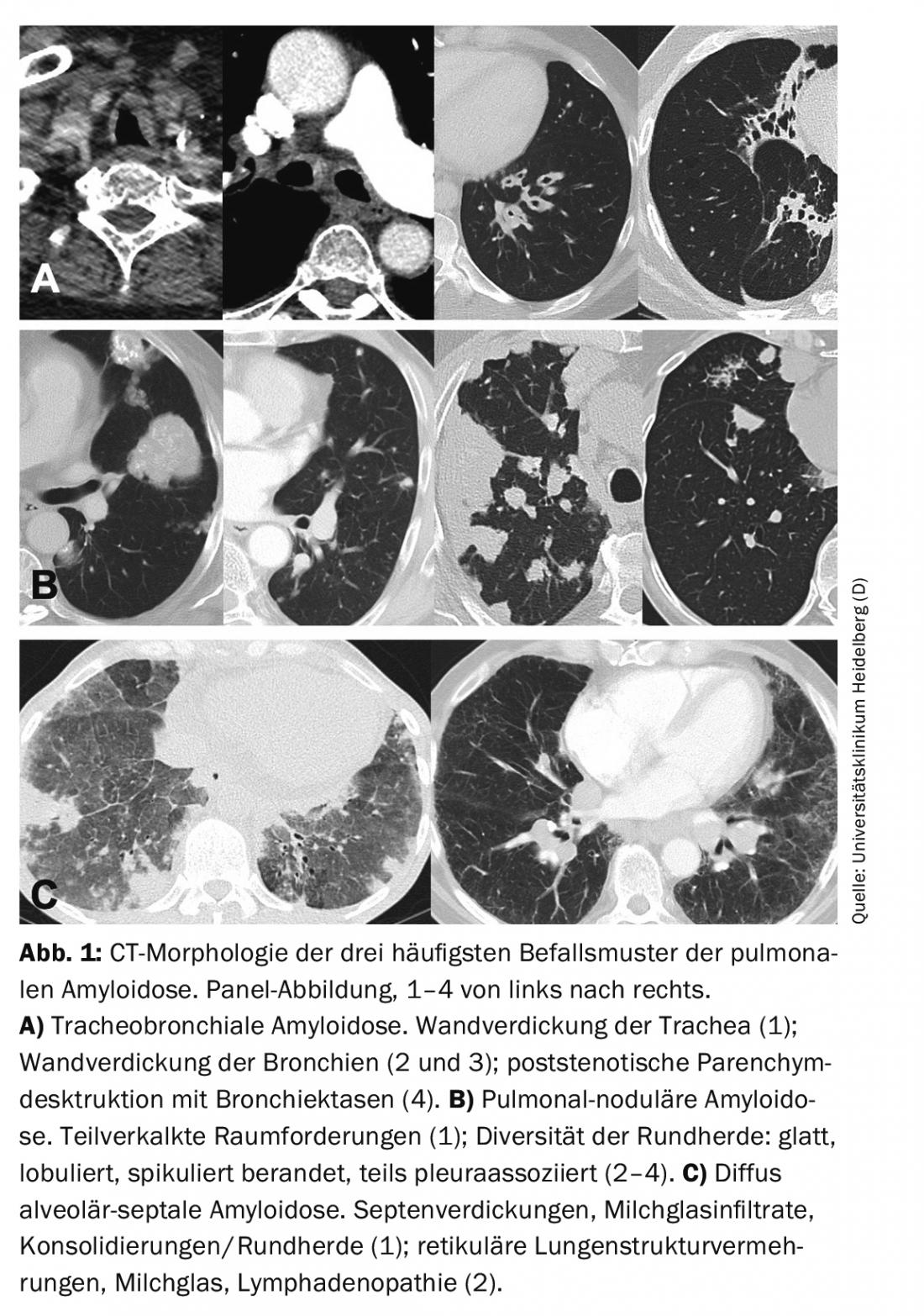
Depending on the site of amyloid deposition, 3 characteristic patterns of pulmonary amyloidosis can be distinguished: tracheobronchial (upper/lower airway), pulmonary nodular, and diffuse alveolar-septal.
Tracheobronchial amyloidosis
Patient 1: We saw a 55-year-old female patient at diagnosis in mildly reduced AZ, BMI 23 kg/m2, with preexisting conditions of type 2 diabetes mellitus, arterial hypertension, persistent inhaled nicotine abuse (35 pack-years), and history of chronic bronchitis in childhood. Due to recurrent bronchitis, repeated antibiotic therapy had been given for more than a year. Furthermore, there was a chronic cough.
Externally, partial atelectasis of the middle lobe and 2 possible round foci were seen by x-ray thorax. CT chest with contrast also showed signs of atelectasis of the middle lobe with very subtle whitish thickening of the middle lobe bronchus, even more subtle of the upper lobe bronchus on the right. Furthermore, there was a concomitant lymphadenopathy on the right hilum. The findings were considered to be a mass with constriction of the right middle lobe bronchus and a lymph node metastasis until proven otherwise. Bronchoscopy with trial excision was performed, revealing histologically distinct interstitial and vascular amyloid deposits type AL lambda. No clonal plasma cells or B cells were detected in the subsequent bone marrow biopsy. No amyloid was found in the material from a rectal biopsy as well as in the adipose tissue aspiration performed at our institution. Echocardiography also revealed no evidence of cardiac involvement of systemic amyloidosis. However, immunofixation electrophoresis revealed monoclonal gammopathy type IgG lambda. We made the diagnosis: local bronchial amyloidosis, type AL lambda, with concomitant monoclonal gammopathy type IgG lambda.
After approximately 1.5 years, an exacerbated infection occurred in terms of progression of bronchial amyloidosis with occlusion of partial bronchi. Endobronchial recanalization and dilatation and thoracic radiotherapy were performed as a single case decision with 20 Gy in 10 fractions. In the following months, the symptoms slowly improved. Over the further observation period of one year known to us, the clinical and radiological condition was stable. There was no evidence of progression of monoclonal gammopathy.
Between April 2000 and October 2019, we saw 12 patients with local amyloidosis of the nasopharynx, 51 pa-tients with local amyloidosis of the larynx, and 31 patients with local amyloidosis of the lower airways at the Heidelberg Amyloidosis Center [6]. Pathophysiologically, this pattern of involvement results in the appearance of multifocal submucosal amyloid plaques on the respiratory epithelium, causing subtotal to total obstruction of the dependent airways. Depending on the localization, proximal, intermediate and distal involvement can be distinguished [14]. Patients often become symptomatic with cough, recurrent respiratory infections, hemoptysis, inspiratory stridor, and hoarseness. Particularly in cases of high-grade proximal stenosis, e.g., subglottic, dyspnea may also be a leading cause, which is often mistakenly attributed to an asthmatic etiology [12].
In pulmonary function diagnostics, tracheobronchial amyloidosis leads to obstruction, especially with proximal involvement [14]. CT morphology may detect wall thickening of the trachea and/or bronchi (Fig. 1). Bronchial stenoses lead to lobar or segmental dystelectasis and may result in reten-tion pneumonia and lung parenchyma destruction. Calcification of the tracheobronchial walls occurs more frequently. In contrast to tracheobronchopathy osteochondroplastica, the posterior wall of the trachea may also be involved [15]. Bronchoscopy reveals irregular whitish deposits that may subtotally to totally constrict the lumen. These are usually diffuse and also involve the posterior wall of the trachea. Lesions are fragile and prone to rebleeding on biopsies. Histologically, amyloid is found in the submucosa and blood vessels, often associated with plasma cells and giant cells. In subtyping, AL is usually detectable [16].
In most cases, tracheobronchial amyloidosis remains confined to the organ and rarely expresses systemic AL amyloidosis [16]. Therefore, systemic chemotherapy is usually not useful for the disease. In cases of severe impairment, e.g., recurrent bronchitis or dyspnea, local therapeutic measures are indicated. Endobronchial ablation of amyloid using lasers or forceps has proven effective. In exceptional cases, focused irradiation is also possible, which is potentially directed against the causative clonal B-cell disease. Pulmonary infections can become life-threatening because of the stenoses and therefore usually require rapid antibiogram therapy.
In the absence of severe hemorrhage or pulmonary infection, the prognosis of local tracheobronchial amyloidosis is very good. In less than 20% of cases, clinically relevant or CT morphologic progression of the disease occurs over the course of 5 years [6].
Pulmonary nodular amyloidosis
Patient 2: We saw a 49-year-old patient in good general condition at diagnosis, BMI 28 kg/m². Except for a condition of inhaled nicotine abuse (15 pack-years, abstinent for 20 years), there were no relevant prior diagnoses.
The initial pneumological presentation was due to a bronchopulmonary infection persisting for weeks as well as fever, night sweats and dyspnea on exertion (weight stable, no cough). Pulmonary function was unremarkable with no diffusion restriction. Eight months before presentation to our center, a CT thorax with contrast was performed. Several, mostly peripherally located, partially calcified compressions were conspicuous in both lungs, which were new or had progressed in size compared to a previous examination 3 years earlier. In addition, multiple cystic lesions were seen bilaterally, but no malignancy-specific space-occupying lesions and no enlarged lymph nodes. Differentially, atypical mycobacteriosis was suspected. A flexible bronchoscopy was performed, which revealed unremarkable endobronchial findings with no direct or indirect tumor signs. No pathogen was detected in bronchoalveolar lavage of the middle lobe, and mycobacteriosis was not detected.
Five months prior to presentation to us, a CT-guided puncture (postinterventional pneumothorax with no need for therapy) of the right lower lobe was performed. This revealed regressively altered interstitial amyloid deposits AL kappa. No clonal plasma cells or B cells and no amyloid were detected in the subsequent bone marrow biopsy. Follow-up by CT shortly before presentation to our center showed constant calcified granulomas bipulmonary.
On presentation to our center, we performed a fat tissue aspiration in which no amyloid was detected. By echocardiography, there was no evidence of cardiac involvement of systemic amyloidosis. By means of immunofixation electrophoresis as well as measurement of free light chains in serum, a low-grade monoclonal gammopathy type IgG kappa could not be excluded.
We made the diagnosis: local pulmonary amyloidosis, type kappa, with V.a. concomitant monoclonal gammopathy type IgG kappa. A follow-up at 6 months showed no progression of the gammopathy and there was still no evidence of systemic AL amyloidosis. We recommended regular pneumological follow-up.
Between April 2000 and October 2019, 63 patients presented to the Heidelberg Amyloidosis Center with local pulmonary nodal amyloidosis [6]. Pathophysiologically, this manifestation results in tumor-like amyloidomas – multifocal deposits of amyloid in the lung parenchyma. This is mostly AL in subtyping. It is currently believed that lymphomas of the mucosa-associated lymphoid tissue (MALT) are causative for most pulmonary nodular amyloidoses [12]. It is often an incidental finding on chest imaging. Less than half of the patients in question show symptoms such as cough, dyspnea, hemoptysis, or bronchopulmonary infections. Therefore, compared with tracheobronchial amyloidosis, diagnosis tends to occur later and the median patient age of patients with pulmonary nodal amyloidosis is higher compared with patients with tracheobronchial amyloidosis (68 years vs. 55 years).
By means of pulmonary function diagnostics, restriction is most likely to occur if the findings are pronounced. CT morphology is usually dominated by a nodular-focal pattern with solitary to multiple space-occupying lesions and/or round foci. The morphology of the foci is diverse with smooth to spiculated borders and optional calcifications and pleural association (Fig. 1). CT morphology cannot differentiate the lesions from malignant or granulomatous disease. Lung cysts, sometimes with associated round foci, and lymphadenopathy may also be part of the varied CT picture [17].
If neoplasia or systemic amyloidosis can be excluded by differential diagnosis, the prognosis of pulmonary nodal amyloidosis is very good. The disease is usually bland, with CT morphologic progression occurring in less than 36% of cases over 5 years [6]. Functionally limiting amyloidomas or cysts can be treated by surgical excision with best possible preservation of the surrounding tissue.
Diffuse alveolar septal amyloidosis.
Patient 3: We saw a 50-year-old female patient in reduced AZ at diagnosis, BMI 22 kg/m². Five months before the first presentation at our center, antibiotic and anti-obstructive therapies were performed for protracted cold symptoms with non-productive cough, which did not lead to an improvement of the symptoms. Furthermore, the patient reported noticing increasing ankle and lower leg edema and recurrent cardiac stuttering and palpitations for about 6 months. There was severe limitation of exercise capacity with dyspnea after only about ½ floor of stair climbing or 150 m of walking on level ground. Until a few weeks ago, there had been significant weight loss (>10% in 6 months); most recently, she had regained weight with clinically pronounced edema. Physical examination revealed macroglossia and marked swelling of the base of the tongue. Anamnestically, recurrent tongue bites occurred in the past months. Furthermore, periorbital hemorrhages were visible, which had been present for approximately one year. When asked, the patient stated that she had received bilateral hand surgery for carpal tunnel syndrome before the onset of pulmonary symptoms.
Native CT thorax showed marked milk glass opacities bilaterally with punctum maximum over the lower lobe segments. Furthermore, multiple peribronchovascular as well as especially subpleural inflammatory changes were present. Hilar lymphoma on the right side was suspected. Diffuse alveoloseptal amyloidosis type AL lambda was detected by transbronchial biopsy. Bone marrow biopsy showed a plasma cell percentage of 80% with lambda light chain restriction. In serum, free lambda light chains were strongly elevated at 7572 mg/l (kappa 8 mg/l).
In summary, we made a diagnosis of systemic AL amyloidosis type lambda. Initial leading symptoms included pulmonary symptoms and pathognomonic soft tissue involvement (macroglossia, periorbital hemorrhage, Z.n. KTS-OP bds). Further organ staging revealed a high-grade cardiac involvement (NT-proBNP 54 749 ng/l, echocardiographically globally still good but longitudinally significantly reduced pump function with a septal thickness of 18 mm) and a renal involvement (proteinuria 8.2 g/d, eGFR 30 ml/min). We recommended comparatively well-tolerated and effective systemic therapy with bortezomib and dexamethasone. Unfortunately, exitus lethalis occurred one month after the initial presentation at our center.
The diffuse alveolar-septal infestation pattern is associated with the clinical picture of interstitial lung disease. With rare exceptions, these patients have systemic AL amyloidosis as their underlying disease [12]. Histopathologically, amyloid deposits are found in the vascular walls and alveolar septa, which can impede gas exchange and lead to pulmonary arterial hypertension. The leading symptom is dyspnea. Pulmonary symptoms are often difficult to assess clinically against the background of usually dominant cardiac disease in systemic amyloidosis.
Restriction and impaired CO diffusion capacity are often detectable by pulmonary function diagnostics. Stress leads to inadequate hypoxemia. CT morphology usually reveals diffuse lung structure proliferation such as septal thickening, milky glass infiltrates, and micronodules (Fig. 1). In addition, space-occupying lesions/consolidations, lung cysts, and lymphadenopathy may be present [17].
Evidence of alveolar-septal amyloidosis of the lung should prompt extensive workup for the presence of systemic AL amyloidosis. Rarely, the pulmonary nodal or tracheobronchial pattern of involvement may also be associated with systemic amyloidosis [6,17]. If a mixture of infestation patterns is present, a systemic course is even very likely [16]. Systemic AL amyloidosis should therefore always be clarified as a differential diagnosis in cases of suspected local pulmonary amyloidosis, especially in the presence of concomitant monoclonal gammopathy.
If no systemic amyloidosis can be detected, close follow-up is of great importance. In the presence of systemic AL amyloidosis, there is an indication for systemic chemotherapy. The choice of chemotherapy regimen here depends largely on the extent of organ involvement. For example, high-dose chemotherapy with autologous stem cell transplantation is no longer considered for patients with significant cardiac involvement or a CO diffusion capacity <50%.
In the setting of systemic amyloidosis, pleural effusions are common, usually in the form of transudate and in up to 1/3 of cases as exudate [12]. If pleural effusions in systemic amyloidosis are refractory to maximal diuretic therapy and pleural punctures, pleural involvement should be considered [12].
Differential diagnosis of local pulmonary amyloidosis versus systemic AL amyloidosis.
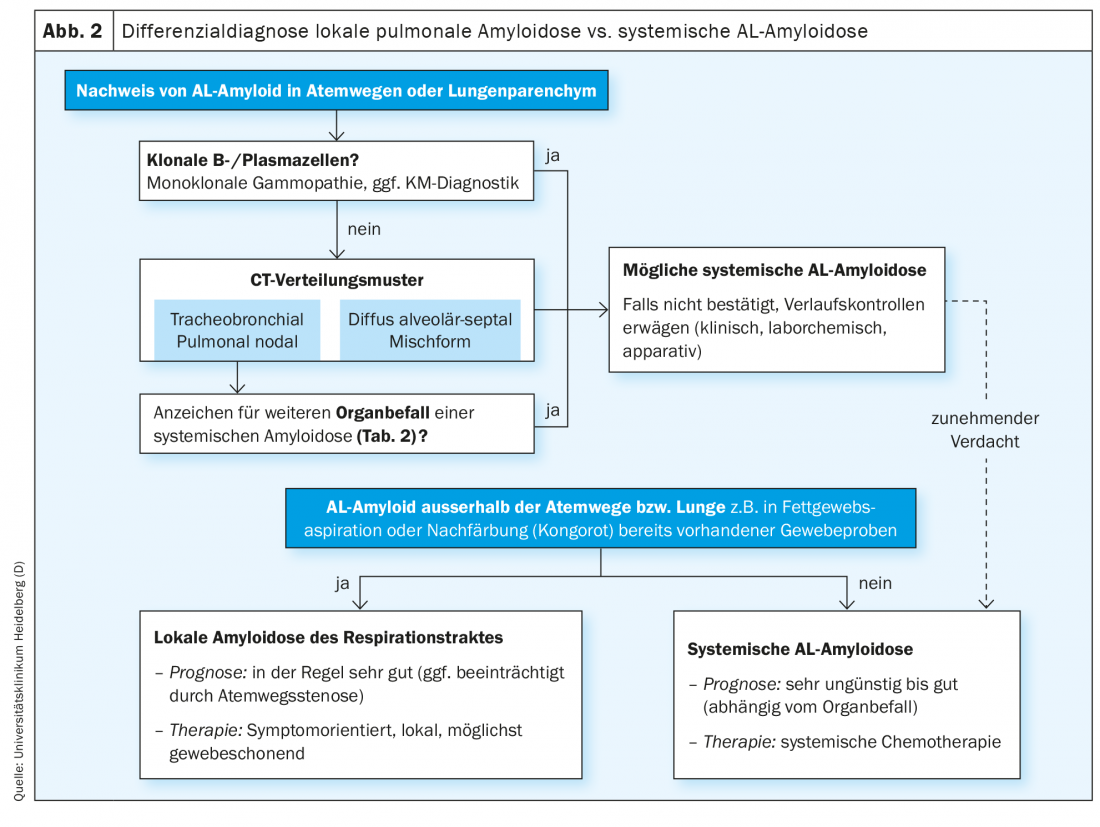
Most histologically confirmed pulmonary amyloidoses have AL subtyping. As described above, most cases are local amyloidoses with a very good prognosis. However, since any pattern of pulmonary amyloidosis can also be associated with a systemic course of amyloidosis, the differential diagnosis of local versus systemic AL amyloidosis is of particular importance, especially in newly confirmed pulmonary AL amyloidosis (Fig. 2) .
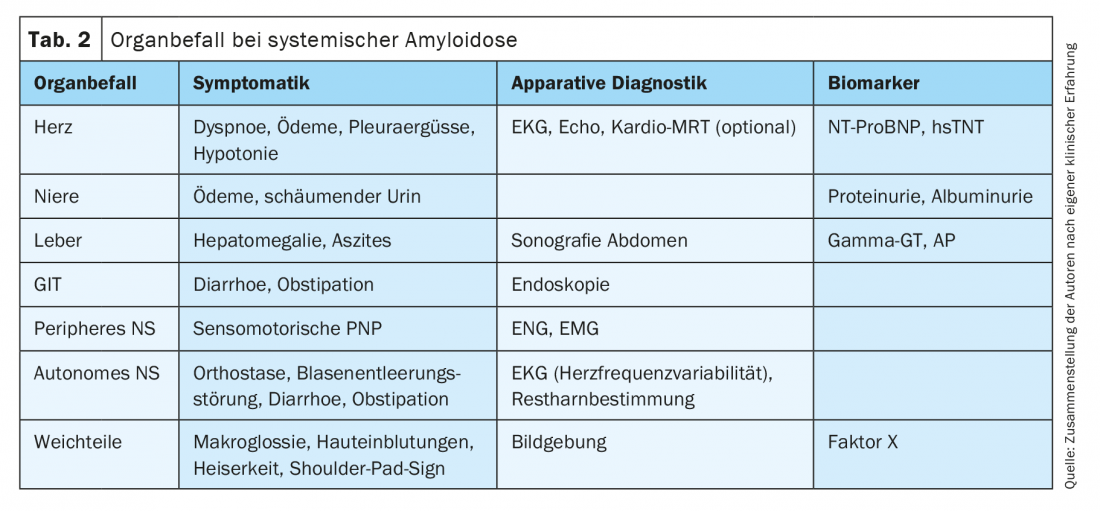
A detailed history and specific questioning about concomitant symptoms may provide clues to another affected organ system in the context of systemic AL amyloidosis (Table 2) . Syncope, arterial hypotension, and pulmonary unexplained exertional dyspnea may indicate cardiac involvement. Renal infestation usually manifests as glomerular damage with protein loss syndrome; less frequently, renal vascular infestation leads directly to renal dysfunction. The constant loss of protein can lead to weight loss in addition to foamy urine, and in the course most patients present with edema. Infestation of the gastrointestinal tract may result in diarrhea or bleeding. The liver is rarely clinically leading affected. However, coagulation disorders are frequently observed due to binding of coagulation factor X by amyloid, even in the setting of marked soft tissue involvement. Pathognomonic for systemic AL amyloidosis are macroglossia as well as periorbital hemorrhages. Very suspicious is also a carpal tunnel syndrome, especially on both sides, or the condition after KTS surgery, since the retinaculum flexorum of the wrist is a typical and early site of deposition of amyloidogenic proteins [18–20]. Peripheral nervous system involvement may manifest as paresthesias, impaired thermesthesia, and attenuated muscle intrinsic reflexes, among other symptoms. Severe orthostatic dysregulation as well as constipation indicate autonomic nervous system involvement.
Comprehensive instrumental organ staging includes vital signs, ECG (peripheral low voltage in cardiac involvement, impaired heart rate variability in autonomic neuropathy), echocardiography (impaired longitudinal function, thickened septum in cardiac involvement), and liver sonography, among others. (Tab. 2). In addition, laboratory biomarkers are the most important indicators of organ damage in the setting of histologically proven amyloidosis (Tab. 2).
The search for a possibly present monoclonal gammopathy is of central importance for the differential diagnosis between systemic and local AL amyloidosis. To ensure sufficient sensitivity, this should be done by combining immunofixation electrophoresis in serum and urine and measurement of free light chains in serum. More than 1/3 of patients with local AL amyloidosis have mostly minor monoclonal gammopathy, but only about half of these cases correspond to the AL deposition subtype (kappa or lambda) [6]. In addition, approximately 22% of patients with local pulmonary AL amyloidosis have concomitant autoimmune disease, such as Sjögren’s syndrome [6]. These observations suggest that patients with local AL amyloidosis have an increased underlying tendency to form multiple B-cell clones, although the pathophysiological background in this regard is still unknown. Monoclonal gammopathy corresponding to the subtype of AL deposits may be an expression of the small local B-cell clone underlying local amyloidosis [4,6,11], but may also indicate the presence of systemic AL amyloidosis and should therefore usually be clarified by bone marrow diagnostics. In the case of systemic AL amyloidosis, clonal B or plasma cells are usually detectable in the bone marrow with light chain restriction corresponding to the subtype of AL deposition. In contrast, if the monoclonal gammopathy or clonal B/plasma cell population does not correspond to the AL deposition subtype, there is no increased risk of systemic AL amyloidosis.
Amyloidoses confined to the lungs are more likely to be associated with inhaled nicotine abuse than systemic AL amyloidosis [16,17]. The cause of this association is still unclear, but could possibly be explained by an increased incidence of lymphoma due to noxious agents.
Take-Home Messages
- Pulmonary amyloidosis is a very rare disease that can be divided CT morphologically into 3 major patterns of involvement: tracheobronchial, pulmonary nodal, and interstitial or alveolar-septal.
- Affected patients are often asymptomatic (especially pulmonary nodal) and clinically symptomatic infestation can usually be well controlled with local measures.
- In local pulmonary amyloidosis (more common), the long-term prognosis is usually very good, but local progressions are common.
- However, any pattern of pulmonary amyloidosis may also reflect systemic AL amyloidosis with a potentially very unfavorable prognosis.
- Especially in the presence of a monoclonal gammopathy, an alveolar-septal pattern of infestation or symptoms suggestive of systemic infestation, a systemic AL amyloidosis must be clarified as a differential diagnosis.
Literature:
- Merlini G, Bellotti V: Molecular mechanisms of amyloidosis. N. Engl. J Med 2003; 349(6): 583-596.
- Benson MD, Buxbaum JN, Eisenberg DS, et al: Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018; 25(4): 215-219.
- Kyle RA, Linos A, Beard CM, et al: Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992; 79(7): 1817-1822.
- Stuhlmann-Laeisz C, Schönland SO, Hegenbart U, et al: AL amyloidosis with a localized B cell neoplasia. Virchows Arch 2019; 474(3): 353-363.
- Meijer JM, Schonland SO, Palladini G, et al: Sjögren’s syndrome and localized nodular cutaneous amyloidosis: coincidence or a distinct clinical entity? Arthritis Rheum 2008; 58(7): 1992-1999.
- Basset M, Hummedah K, Kimmich C, et al: Localized immunoglobulin light chain amyloidosis: novel insights including prognostic factors for local progression. Am J Hematol 2020; 1158-1169.
- Veelken K, Hegenbart U, Schönland SO, Blank N.: Local and systemic light chain amyloidosis in patients with rheumatic diseases. Z Rheumatol 2020; 660-668.
- Dittrich T, Benner A, Kimmich C, et al: Performance analysis of AL amyloidosis cardiac biomarker staging systems with special focus on renal failure and atrial arrhythmia. Haematologica 2019; 104(7): 1451-1459.
- Dittrich T, Bochtler T, Kimmich C, et al: AL amyloidosis patients with low amyloidogenic free light chain levels at first diagnosis have an excellent prognosis. Blood 2017; 130(5): 632-642.
- Kimmich C, Schönland S, Kräker S, et al: Amyloid in bone marrow smears in systemic light-chain amyloidosis. Amyloid 2017; 24(1): 52-59.
- Baumgart J-V, Stuhlmann-Laeisz C, Hegenbart U, et al: Local vs. systemic pulmonary amyloidosis-impact on diagnostics and clinical management. Virchows Arch 2018; 473(5): 627-637.
- Milani P, Basset M, Russo F, et al: The lung in amyloidosis. Eur Respir Rev 2017; 26(145): 170046.
- Ussavarungsi K, Yi ES, Maleszewski JJ, et al: Clinical relevance of pulmonary amyloidosis: an analysis of 76 autopsy-derived cases. Eur Respir J 2017; 49(2): 1602313.
- O’Regan A, Fenlon HM, Beamis JF, et al: Tracheobronchial amyloidosis. The Boston University experience from 1984 to 1999. Medicine (Baltimore) 2000; 79(2): 69-79.
- Czeyda-Pommersheim F, Hwang M, Chen SS, et al: Amyloidosis: Modern Cross-sectional Imaging. Radiographics 2015; 35(5): 1381-1392.
- Rech JS, Arnulf B, de Margerie-Mellon C, et al: Lower respiratory tract amyloidosis: presentation, survival and prognostic factors. A multicenter consecutive case series. Am J Hematol 2019; 94(11): 1214-1226.
- Brandelik SC, Heussel CP, Kauczor HU, et al: CT features in amyloidosis of the respiratory system – Comprehensive analysis in a tertiary referral center cohort. Eur J Radiol 2020; 129: 109123.
- Kelly JJ: Peripheral neuropathies associated with monoclonal proteins: A clinical review. Muscle & Nerve 1985; 8(2): 138-150.
- Haan J, Peters WG: Amyloid and peripheral nervous system disease. Clinical Neurology and Neurosurgery 1994; 96(1): 1-9.
- Sperry BW, Reyes BA, Ikram A, et al: Tenosynovial and Cardiac Amyloidosis in Patients Undergoing Carpal Tunnel Release. J Am Coll Cardiol 2018; 72(17): 2040-2050.
InFo PNEUMOLOGY & ALLERGOLOGY 2020; 2(4): 11-17.