
In hematology, the annual meeting of the European Hematology Association is one of the dates in the annual schedule. The association promotes excellence in patient care, research and education in hematology. Accordingly, the congress conveys the latest and most innovative findings and research results concerning hematological diseases. This will span clinical research and practice, basic and translational research, and the latest approaches to diagnosis and treatment.
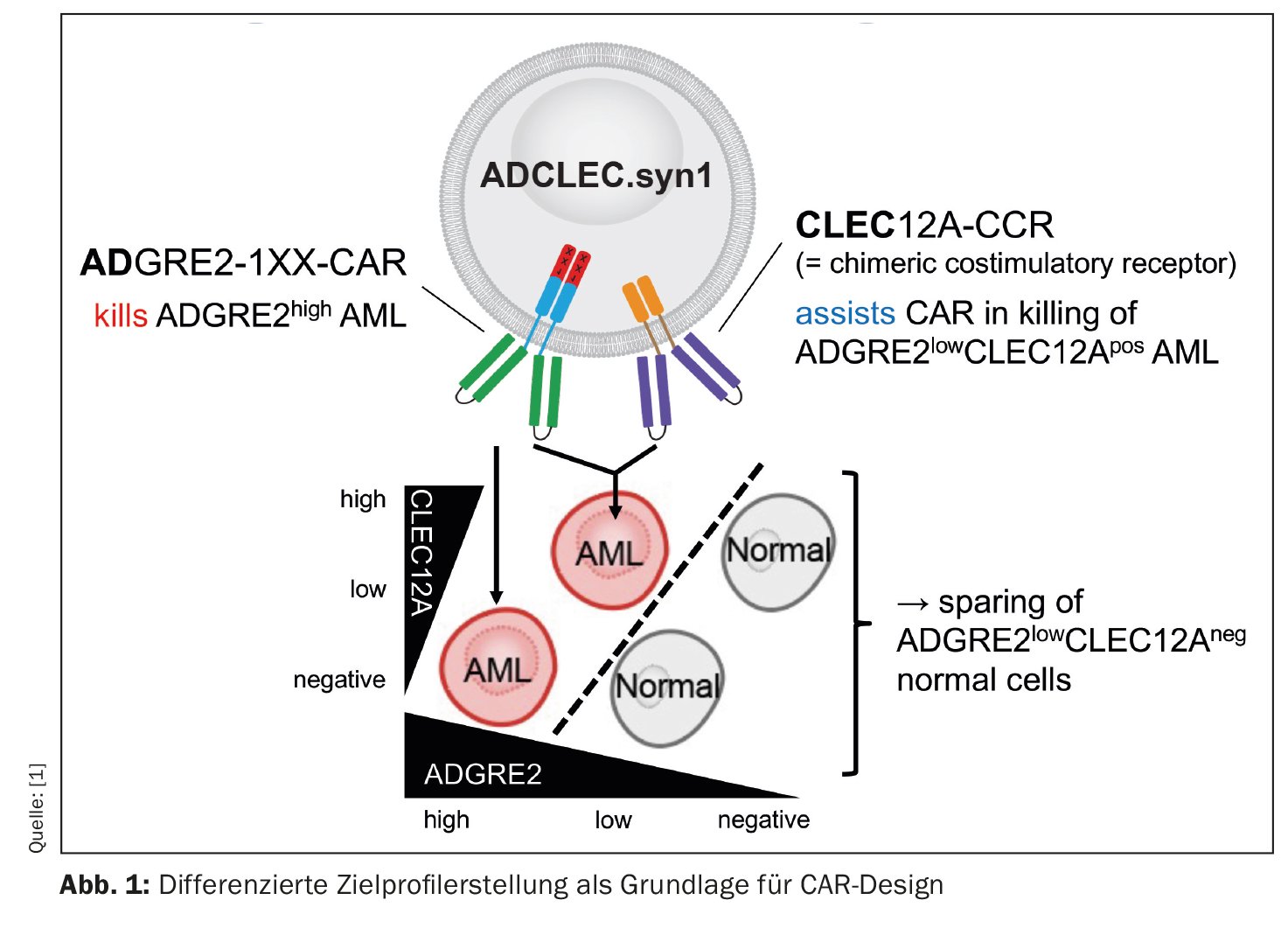
A novel CAR therapy for acute myeloid leukemia (AML) is being addressed by a group of researchers whose preclinical results are encouraging [1]. The concept, called ADCLEC.syn1, uses cooperative receptors targeting ADGRE2 and CLEC12A. In this way, AML should be eliminated while minimizing hematologic toxicities. This has been demonstrated by a comprehensive series of in vivo efficacy and toxicity models. CAR therapies for AML face obstacles due to clonal heterogeneity and similarity to normal early hematopoiesis, which can lead to antigen escape and hematologic toxicities. The current study examined quantitative expression of surface targets in AML and normal tissues to determine therapeutic windows that can be exploited by novel combinatorial CAR designs. Thus, ADCLEC.syn1 was developed, a novel combinatorial CAR therapy that co-targets ADGRE2 and CLEC12A for selective elimination of AML cells with low levels of ADGRE2 and sparing of normal hematopoietic stem and progenitor cells (Fig. 1). Investigators correlated target antigen expression with ADCLEC.syn1 and CD33-CAR T cell efficacy using AML xenografts. The results showed that ADCLEC.syn1 induced durable remission in several human AML cell lines representative of the phenotypes of relapsed/refractory AML patients. However, mice that received AML grafts and were reconstituted with normal human hematopoietic cells responded only to ADCLEC.syn1 but not to CD33-CAR. These results highlight the importance of quantitative CAR target profiling in AML. ADCLEC.syn1 is now being evaluated in a first-in-human Phase I clinical trial for relapsed/refractory AML.
Progress at APL
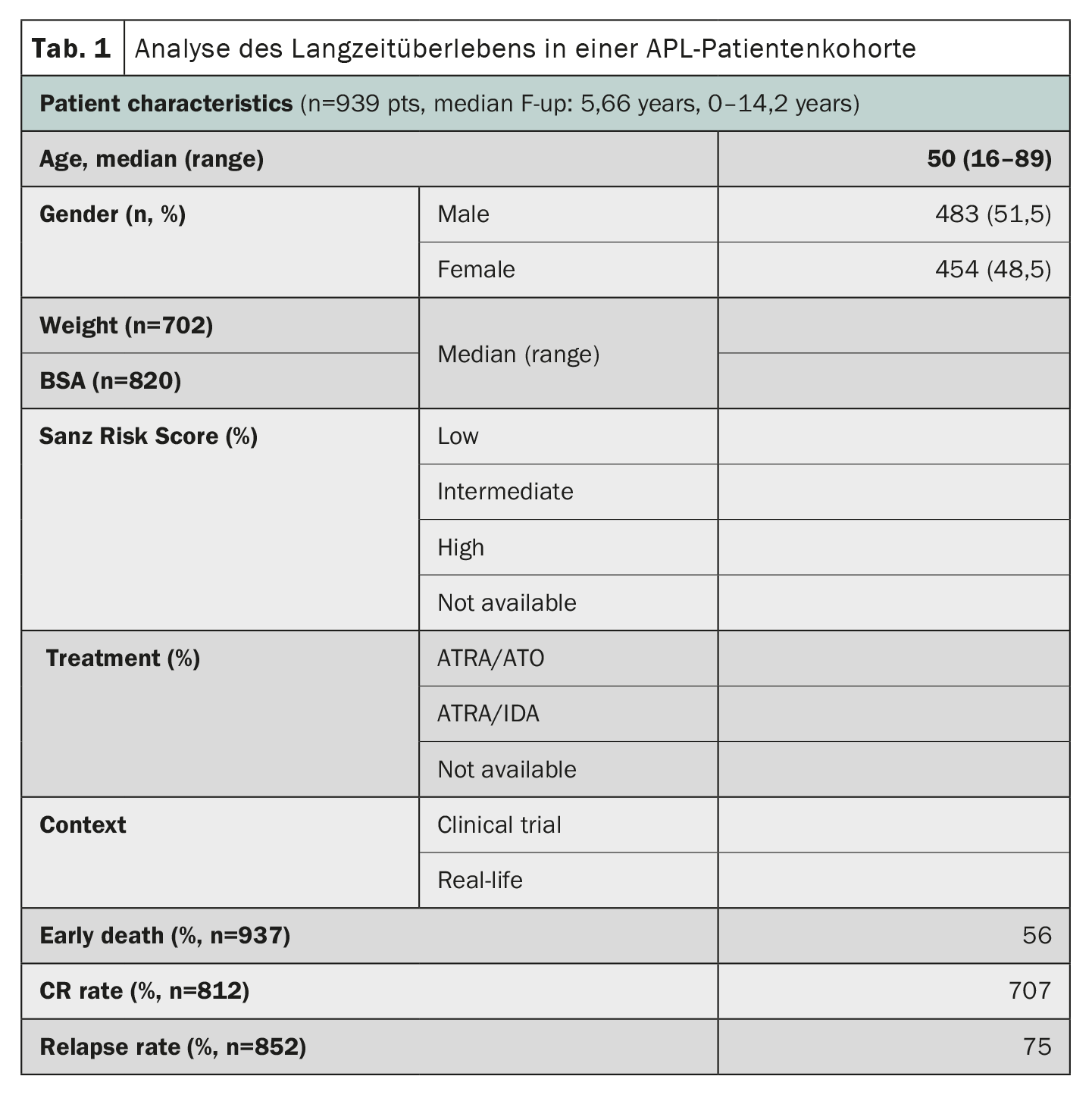
Acute promyelocytic leukemia (APL), once considered one of the most rapidly fatal forms of acute myeloid leukemia, has shown remarkable progress in treatment outcomes. A study using data from the HARMONY registry with a large cohort of patients confirmed that combination therapy of all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) resulted in 10-year overall survival rates of 85-92% in patients with APL [2]. The HARMONY registry includes 1868 patients with APL, from two clinical trials and national registries in six countries, diagnosed between 2007 and 2020. Of these, 937 patients met data quality requirements and were included in the present analysis. Data were harmonized using an Observational Medical Outcomes Partnership Common Data Model and registered in the HARMONY Big Data Platform. The results of the analysis showed that patients treated with the ATRA-ATO regimen had a 10-year overall survival (OS) rate of 92%, compared with 75% for patients treated with the ATRA-idarubicin (AIDA) regimen (Table 1). The survival benefit was the same in the different risk groups defined by the Sanz risk score. Age also played an important role in survival, with younger patients (<50 years) having better outcomes. However, the rate of early death (<30 days after diagnosis) was similar in both groups (3.4%-5.7%). Overall, these results in a large international patient cohort confirm the significant survival benefit of chemo-free ATRA-ATO therapy for APL patients, regardless of their risk profile, and provide valuable insights for the treatment of APL.
Functional healing in TDT?
Initial interim results inspire confidence [3]: A phase III trial of exagamglogenic autotemcel (exa-cel), a non-viral cell therapy, is raising hope for a one-time functional cure for patients with transfusion-dependent β-thalassemia (TDT). Significant results were demonstrated in transfusion independence, improved hemoglobin levels, and quality of life. Exa-cel reactivates fetal hemoglobin (HbF) synthesis by ex vivo CRISPR/Cas9 gene editing, targeting the BCL11A gene in autologous CD34+ hematopoietic stem and progenitor cells. Of the 48 TDT patients who received exa-cel, 27 were evaluable for study end points at the pre-specified interim analysis. Those who were transfusion-independent for ≥12 months had a mean time to last transfusion of 37 days after exa-cel infusion and remained transfusion-free for 12.1-40.7 months. Hemoglobin levels and edited BCL11A alleles in bone marrow CD34+ cells and peripheral nucleated blood cells remained stable over time.
Significant improvements in quality of life and successful neutrophil and platelet engraftment were also observed in all patients in the study, underscoring the efficacy of the therapy. The safety profile of exa-cel was consistent with the busulfan-based myeloablative conditioning regimen and autologous transplantation procedures, with manageable adverse events. All serious adverse events resolved and there were no deaths, study discontinuations, or malignancies.
Uncontrolled erythrocytosis in polycythemia vera.
The REVIVE trial evaluated the efficacy of rusfertide, a novel hepcidin mimetic, in patients with polycythemia vera (PV) [4]. Rusfertide inhibits red blood cell production in PV patients by limiting iron availability. The study used a synthetic protein similar to hepcidin, which is normally produced by the liver and regulates iron trafficking, to treat PV-associated erythrocytosis (excessive red cell production). The 12-week randomized withdrawal phase of the study met the primary endpoint and demonstrated the high efficacy of rusfertide in controlling erythrocytosis, a key feature of PV that increases the risk of thromboembolic and cardiovascular complications.
The phase II study enrolled patients diagnosed with PV according to the 2016 WHO criteria and who required an excessive number of therapeutic phlebotomies (TP) in patients treated with TP alone or with cytoreduction agents (CYTO). Rusfertide, administered subcutaneously once weekly, was added to the previous PV therapy. During the withdrawal phase, patients were randomized to continue rusfertide therapy for 12 weeks or to receive placebo. Data from the randomized withdrawal phase demonstrated the superior efficacy of rusfertide compared with placebo. Patients showed a statistically significant response rate (TP-free) of 69.2% versus 18.5% in the placebo group. In addition, rusfertide therapy was associated with sustained control of hematocrit (HCT) compared with placebo. The TP-free rate in patients reached 92.3%. Treatment was generally well tolerated, with most adverse events being injection site reactions of mild to moderate severity that decreased with continued treatment.
The positive results demonstrate the efficacy and tolerability of rusfertide as a highly effective therapy for uncontrolled erythrocytosis and associated symptoms in PV and represent a significant advance in the treatment of this malignant myeloproliferative neoplasm. The agent offers a novel approach based on a hormone mimetic that selectively targets uncontrolled erythrocytosis, resulting in sustained and durable HCT control and improvement of PV-related symptoms.
Anemia treatment in LR-MDS
The phase III COMMANDS trial showed promising results in the treatment of anemia associated with lower-risk myelodysplastic syndromes (LR-MDS) [5]. In a pre-planned interim analysis, luspatercept demonstrated significant clinical benefits in ESA-naïve LR-MDS patients compared to standard treatment with epoetin alfa. These findings have the potential to change the treatment landscape for LR-MDS patients who rely on transfusions. LR-MDS patients suffering from chronic anemia experience increased morbidity, iron overload, and reduced survival. The current standard of care, erythropoiesis-stimulating agents (ESAs), achieve suboptimal results in these patients. The interim analysis evaluated the efficacy and safety of luspatercept and epoetin alfa in 356 ESA-naive, transfusion-dependent LR-MDS patients. The primary end point of the study was the achievement of red blood cell transfusion independence (RBC-TI) for ≥12 weeks with a concomitant mean hemoglobin increase of at least 1.5 g/dL during the first 24 weeks. 59% of luspatercept-treated patients achieved RBC-TI and concomitant hemoglobin increase compared with 31% in the epoetin-alfa group. Clinical benefit was observed in all subgroups. Achievement of secondary endpoints also favored luspatercept, including hematologic improvement. Patients with specific MDS-associated gene mutations, such as SF3B1, SF3B1a, ASXL1, and TET2, had an above-average response to luspatercept, regardless of their overall mutation burden. Luspatercept had a favorable safety profile, with mild to moderate treatment-related adverse events. The overall mortality rate was similar in the luspatercept and epoetin alfa groups.
Treat CLL effectively
The CLL14 trial, an investigation into the treatment of chronic lymphocytic leukemia (CLL), has provided recent findings on the long-term outcomes of patients treated with venetoclax-obinutuzumab (Ven-Obi) [6]. The results demonstrate sustained efficacy and safety and could establish Ven-Obi as a preferred treatment option for CLL patients, including those with concomitant disease. In this ongoing study, 432 previously untreated CLL patients were randomly assigned to treatment with Ven-Obi or chlorambucil-obinutuzumab (Clb-Obi). After a median follow-up of 76.4 months, Ven-Obi showed better progression-free survival (PFS) than Clb-Obi (median PFS 76.2 vs. 36.4 months). Importantly, even at six years, the estimated PFS rate for Ven-Obi was 53.1%, compared with 21.7% for Clb-Obi. The study also showed that Ven-Obi resulted in significantly longer time to next treatment (TTNT) compared with Clb-Obi (6-year TTNT 65.2% vs. 37.1%) (Fig. 2). These positive results were observed in all risk groups, including patients with high-risk CLL features. In addition, Ven-Obi showed an excellent minimal residual disease (MRD) response, with 7.9% of patients with no detectable MRD levels five years after treatment, compared with 1.9% for Clb-Obi. New safety signals were not detected.
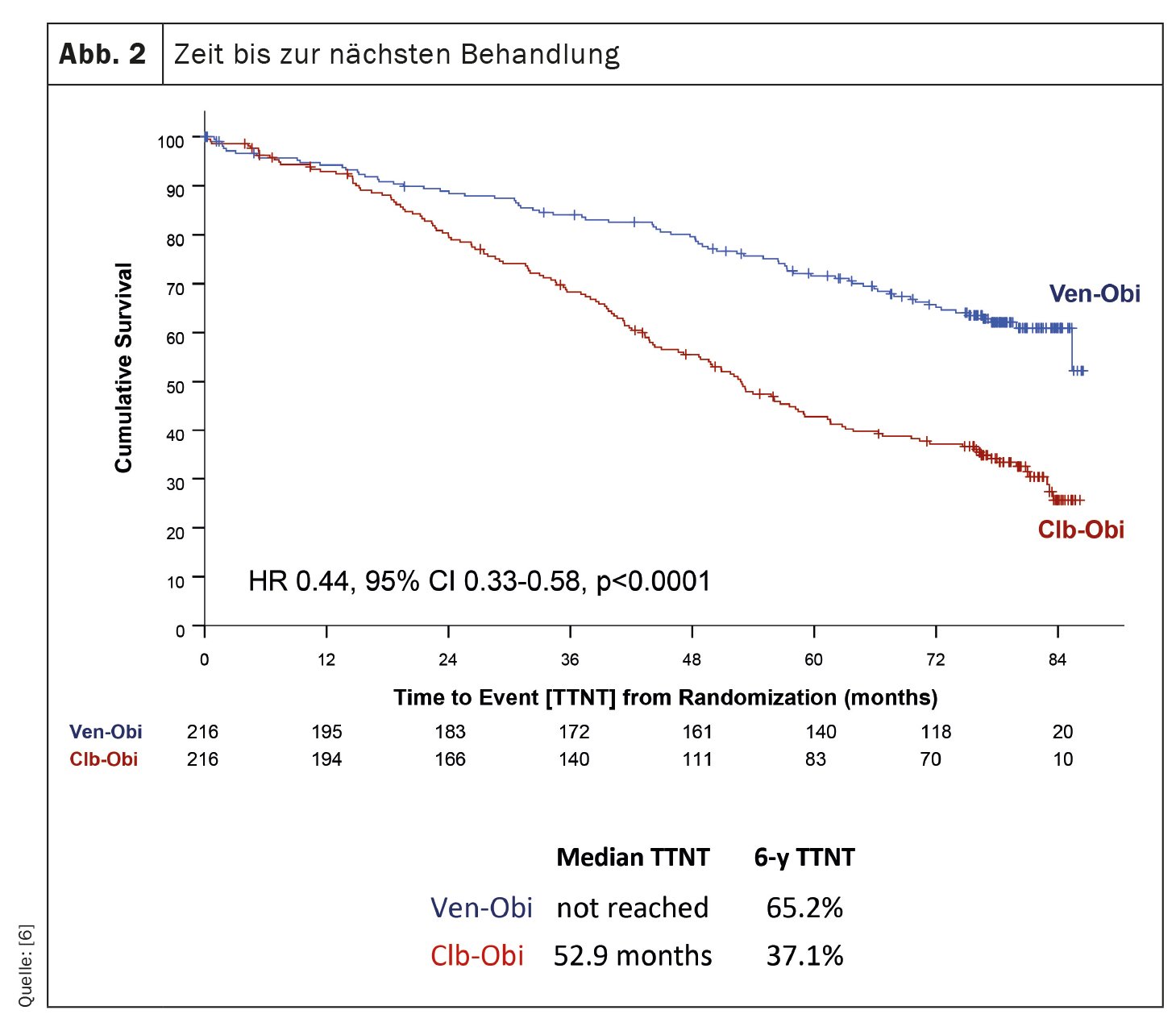
Ven-Obi provides sustained remission, high rates of undetectable minimal residual disease, and extended time to next treatment. More than half of patients are still in remission five years after completing treatment and the majority do not require second-line treatment.
Hereditary hemochromatosis
Homozygosity for the HFE variant C282Y causes hereditary hemochromatosis, which can potentially lead to diabetes, liver disease, and heart disease. A study has now tested whether C282Y homozygosity increases the risk of diabetes, liver disease, and heart disease, even in individuals with normal plasma iron, transferrin saturation, or ferritin [7]. In addition, we investigated whether C282Y homozygotes with diabetes, liver disease, or heart disease have an increased risk of death compared with non-carriers with these diseases. A total of 132 542 consecutive individuals from a general population cohort were genotyped for the HFE C282Y variant and 422 homozygotes were found. Individuals were followed prospectively for up to 27 years. Information on hospital contacts came from the National Patient Register, which covers all Danish hospitals.
During the follow-up period, 17 688 subjects died from any cause, while 7702, 2804, and 21 769 subjects had hospitalizations for diabetes, liver disease, and heart disease, respectively. When C282Y homozygote carriers were compared with noncarriers, the hazard ratios were 1.66 for diabetes, 2.16 for liver disease, and 0.98 for heart disease. Diabetes risk was increased even in C282Y homozygotes with normal iron, transferrin saturation, or ferritin (4.35; 1.81-10.48). C282Y homozygotes with diabetes had a hazard ratio for death from any cause of 1.94 compared with noncarriers with diabetes. The percentage of all deaths in C282Y homozygotes that could theoretically be prevented if a single disease were eliminated was 27.3% for diabetes and 14.4% for liver disease. The findings suggest that the current hemochromatosis treatment strategy, which focuses on lowering ferritin, may inadequately reduce diabetes risk and the risk of dying from diabetes.
European Hematology Association (EHA) Congress 2023
Literature:
- Hauber S, et al.: Novel CAR Therapy for Acute Myeloid Leukemia Targeting ADGRE2 and CLEC12A Shows Favorable Pre-Clinical Outcomes. S104. 10.06.2023. EHA 2023.
- Voso MT, et al.: ATRA-ATO Regimen Provides Significant Survival Advantage for Patients with Acute Promyelocytic Leukemia. S136. 11.06.2023. EHA 2023.
- Locatelli F, et al.: Exagamglogene Autotemcel: A Potential One-Time Functional Cure for Patients with Transfusion-Dependent β-Thalassemia. S270. 11.06.2023. EHA 2023.
- Hoffman R, et al.: Rusfertide Therapy Serves as a Novel Effective Treatment for Uncontrolled Erythrocytosis in Polycythemia Vera. LB2710. 11.06.2023. EHA 2023
- Della Porta MG, et al.: Luspatercept Is Superior to Epoetin Alfa in Treating Anemia in Lower-Risk Myelodysplastic Syndromes. S102. 10.06.2023. EHA 2023.
- Al-Sawaf O, et al.: Long-Term CLL14 Study Confirms Venetoclax-Obinutuzumab as Effective Treatment for Chronic Lymphocytic Leukemia. S145. 09.06.2023. EHA 2023.
- Mottelson M, et al.: Individuals with HFE C282Y Homozygosity and Diabetes habe almost two-fold risk of death compared to non-carriers with diabetes: A prospective study of a 132,542-individual general population cohort. S280. EHA 2023.
InFo ONKOLOGIE & HÄMATOLOGIE 2023; 11(3): 26–28









