Primary immunodeficiencies (PGDs) often manifest in childhood and can lead to significant impairment into adulthood due to delayed diagnosis [1]. Effective therapies exist that can reduce morbidity and allow patients to lead a largely normal life [2].
Primary immunodeficiencies (PGDs) comprise a heterogeneous group of more than 400 genetic defects that cause immune dysfunction, including pathological susceptibility to infection and autoimmune diseases [2]. In Switzerland, the prevalence is estimated to range from 1:10 000 to 1:500 000, depending on the disease and region [3]. However, the number of PGD cases has risen sharply in recent years thanks to diagnostic advances, which is why a significantly higher prevalence is now assumed [2]. By far the largest group of PGDs is antibody deficiency disorders, which are caused by decreased antibody production and affect approximately half of PGD patients [2, 4].
Early clarification can reduce mortality
Pathological susceptibility to infection is an important leading symptom of PID [2]. However, the clinical presentation is very heterogeneous due to the variety of genetic defects involved and also includes autoimmune and autoinflammatory disorders. This heterogeneity, as well as the often difficult differentiation from other diseases, leads to a significant diagnostic latency, with devastating consequences: According to a study of 2212 patients from the European Society of Immunodeficiencies (ESID) registry, the risk of death increases by 1.7% with each year of diagnostic delay [5]. Thus, early detection plays a central role in preventing long-term damage [2]. If two or more of the ten major warning signs are present, a workup for PID is appropriate. In particular, the most common antibody deficiency disease can be easily diagnosed in the office setting by quantifying the immunoglobulins IgM, IgG, IgA, and IgE.
10 warning signs of primary immunodeficiency in children
See Jeffrey Model Foundation: http://downloads.info4pi.org/pdfs/10-Warning-Signs—Generic-Text–2-.pdf |
Treatment options for PGD
Treatment of PID is guided by the underlying pathogenesis (overview in Table 1). Antibody deficiency diseases, which are often associated with pathological susceptibility to infections, are readily treatable with immunoglobulin (Ig) replacement therapy: it compensates for the antibody deficiency and thus reduces the frequency and severity of infections. Ig preparations are purified from pooled plasma from 10 000 to 60 000 healthy donors and injected into patients either intravenously or subcutaneously [3].
Table 1: Overview of treatment strategies in PGD. Adapted from [6]
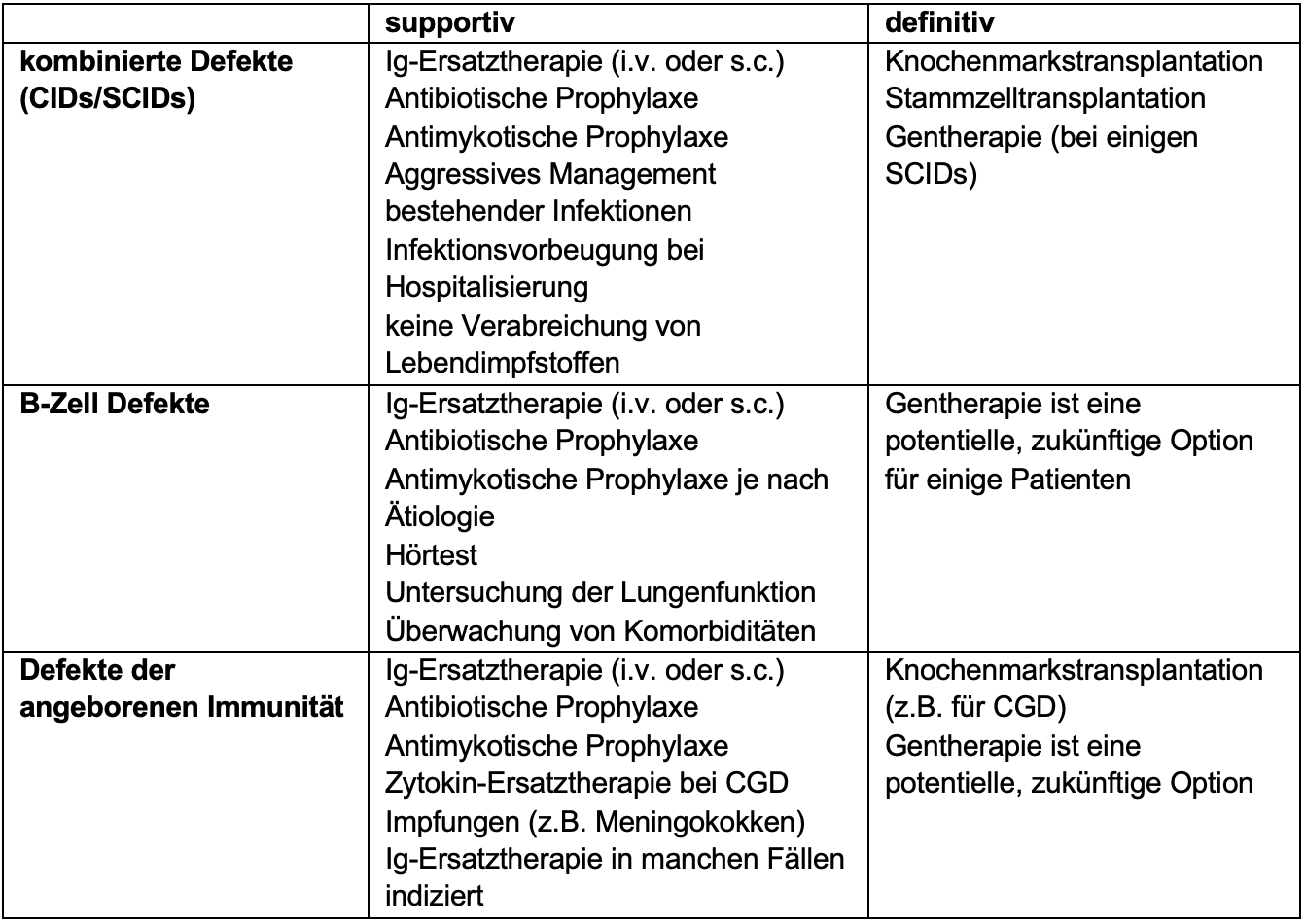
CID: combined immunodeficiency, SCID: severe combined immunodeficiency, i.v.: intravenous; s.c.: subcutaneous, CGD: chronic granulomatosis, Ig: immunoglobulin.
Ig replacement therapy: clear instruction for antibody deficiency diseases
Based on the published data, the German Association of the Scientific Medical Societies (AWMF) has established a guideline for the indication of Ig replacement therapies in antibody deficiency diseases(Table 2) [7]. According to the latter, in addition to pathological susceptibility to infection, an inadequate vaccination response is another essential criterion for the indication of Ig replacement therapy. However, in cases of clinical urgency, Ig replacement therapy should not be delayed because of an investigation of vaccine response.
The efficacy of Ig replacement therapy has been demonstrated in various studies in agammaglobulinemia as well as hypogammaglobulinemia with impaired vaccine response [7]. This also applies to patients with common variable immunodeficiency (CVID), the most common clinically relevant antibody deficiency disease.
Table 2: Data on the benefit of Ig replacement therapy for specific antibody deficiency diseases. Adapted from [7]
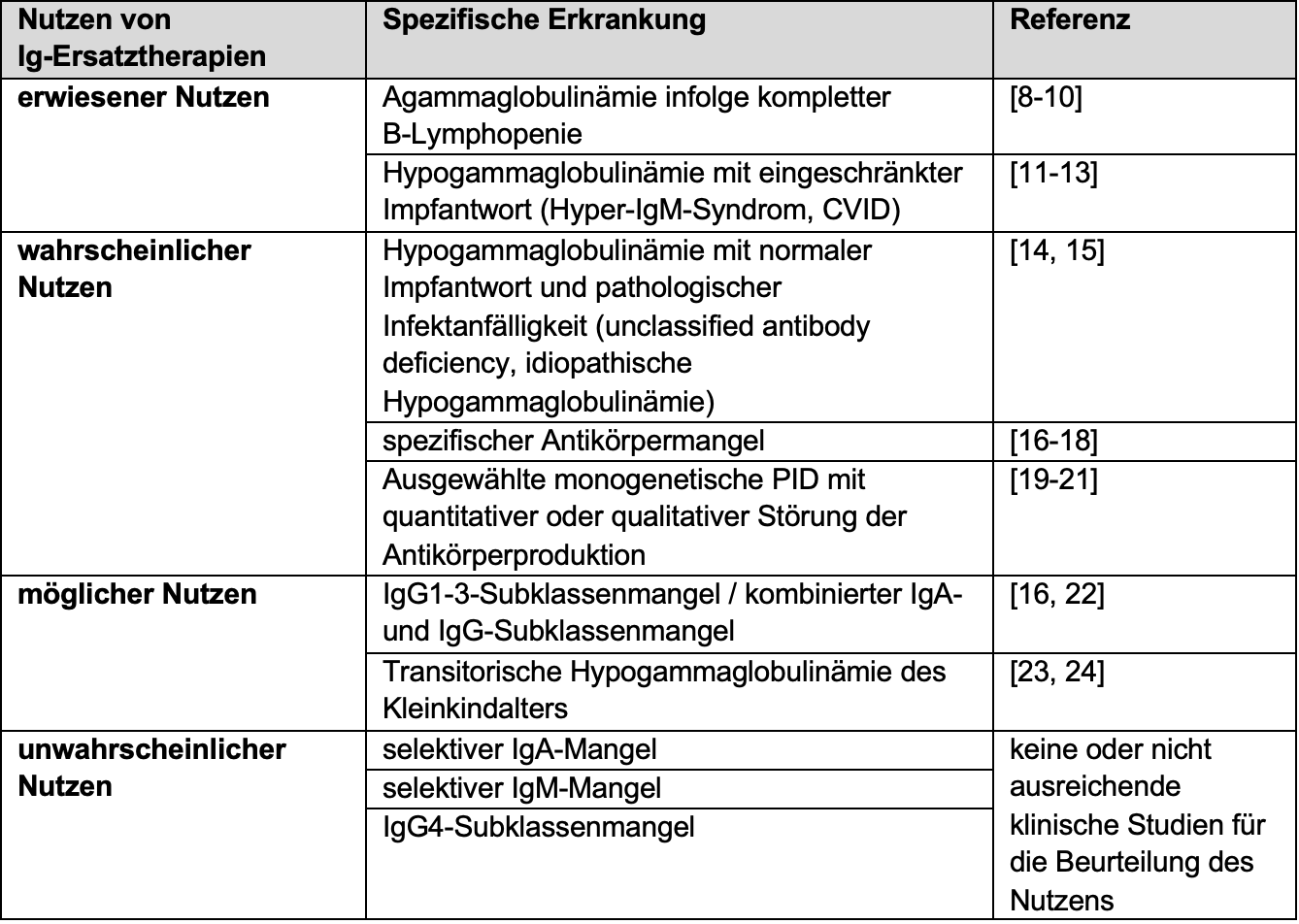
Ig: immunoglobulin, CVID: variable immunodeficiency syndrome
Immunomodulatory approaches for noninfectious manifestations.
The management of autoimmune and autoinflammatory manifestations is a major challenge and is guided by the already established therapies of the respective disciplines. In particular, CVID patients often suffer autoimmune diseases, especially immunocytopenias, which are well treatable with glucocorticoids and rituximab in relapse [2]. High-dose Ig replacement therapies also showed immunomodulatory efficacy in autoimmune thrombocytopenias and are increasingly used in autoimmune manifestations [25]. The AWMF recommends Ig replacement therapy in CVID patients even in the absence of pathologic susceptibility to infection [7].
Stem cell transplantation and gene therapy for combined defects.
Antibody deficiency diseases with additional cellular immunodeficiency may require hematopoietic stem cell transplantation, as therapy with immunoglobulins and/or immunomodulatory agents alone is often insufficient [2]. Gene therapy approaches, in which autologous stem cells are re-transplanted after replacement of the defective gene, have already been shown to improve survival in some combined immunodeficiencies and are currently also being investigated for their efficacy in B-cell defects and defects of innate immunity [6].
Vaccinations reduce the risk of infection
In antibody-deficient diseases with additional impairment of cellular immunity, permanent antibiotic prophylaxis against opportunistic pathogens may be necessary. In addition, PID patients should receive the recommended vaccinations because, although their effectiveness in these patients cannot be accurately predicted, they can increase protection against dangerous infectious diseases. However, live vaccines are prohibited in PID [3].
Conclusion
Timely diagnosis is essential to adequately treat PID and prevent long-term sequelae [2]. A large number of studies show that Ig replacement therapies are effective in antibody deficiency disorders, the most common form of PID [7]. However, stem cell transplantation may be required for combined defects [2]. As a supportive measure, vaccination can increase protection against dangerous infectious diseases [3].
Literature
This text was produced with the financial support of Takeda Pharma AG.
C-ANPROM/CH/CUVI/0013 09/2021
Article online since 05.10.2021