La detección precoz, la estratificación del riesgo y una terapia adaptada para la esclerosis sistémica (SSc) son de gran importancia para poder contrarrestar a tiempo las limitaciones relacionadas con la enfermedad y las manifestaciones potencialmente mortales. La enfermedad pulmonar intersticial (EPI) es una manifestación orgánica de la SSc con una mortalidad significativa. Además de los inmunosupresores, el fármaco antifibrótico nintedanib también está disponible en Suiza desde hace algún tiempo y el tocilizumab se aprobó recientemente en EE.UU. basándose en los resultados positivos de la fase III.
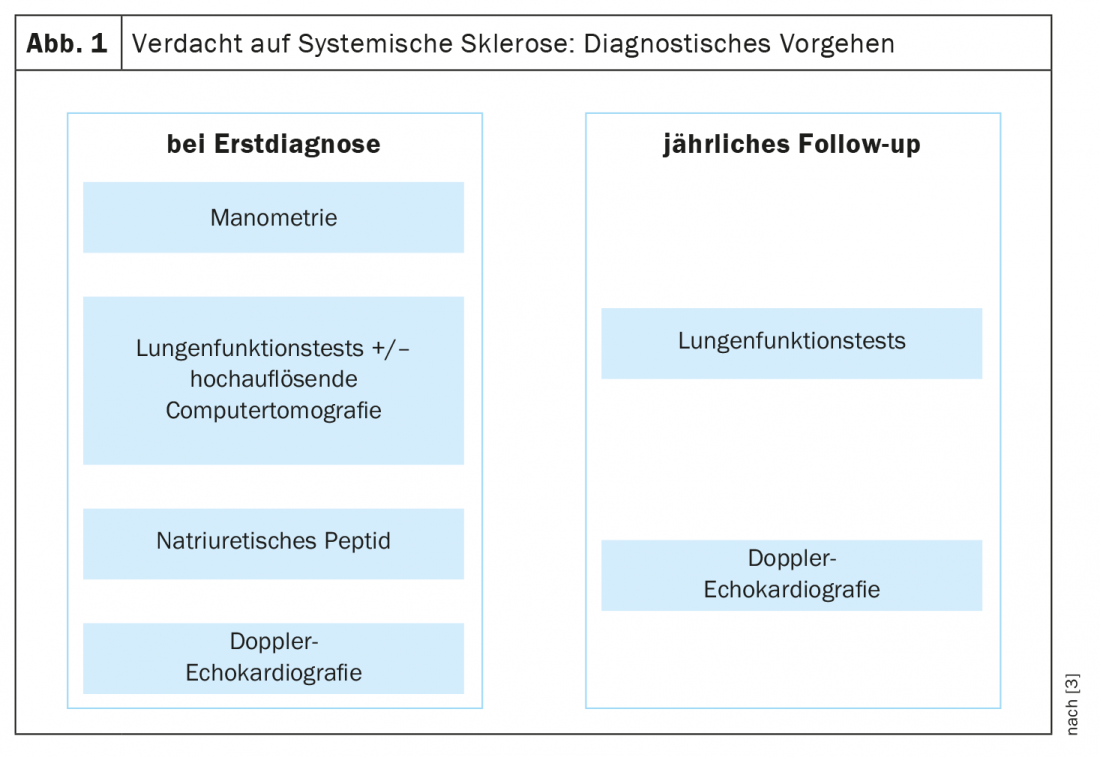
Para el diagnóstico precoz de la esclerosis sistémica se establecieron hace unos años los criterios VEDOSS (“Very Early Diagnosis of Systemic Sclerosis”) [1,2]. El síndrome de Raynaud, la hinchazón de los dedos (“dedos hinchados”), los cambios en los capilares del pliegue ungueal y la detección de autoanticuerpos antinucleares (ANA) son predictivos de la esclerosis sistémica (SSc), explicó la Dra. Hanna Grasshoff, de la Clínica de Reumatología e Inmunología Clínica del Hospital Universitario Schleswig-Holstein de Lübeck [1]. Los criterios de clasificación establecidos en 2013 por el Colegio Americano de Reumatología (ACR) y la Liga Europea contra el Reumatismo (EULAR) siguen siendo válidos [1,16]. Los ANA más comunes asociados a la SSc incluyen el anticéntrico-Ak (ACA) y el anti-topoisomerasa-Ak (ATA, Scl70). Los resultados empíricos actuales confirman que los criterios VEDOSS son adecuados para la estratificación del riesgo de los pacientes. Así lo demostró también un estudio publicado en 2021 en la revista European Journal of Internal Medicine [3]. El ponente resume el procedimiento de diagnóstico del siguiente modo (Fig. 1) : “Los pacientes son examinados con manometría esofágica, pruebas de función pulmonar y, si es necesario, tomografía computerizada de alta resolución si hay anomalías, así como péptido natriurético y ecocardiografía Doppler” [1]. Un ECG adicional puede ser útil [1]. Se recomiendan pruebas anuales de función pulmonar y ecocardiografías Doppler durante el curso. En cuanto a la hipertensión arterial pulmonar, incluida la SSc-PAH, se ha publicado una nueva directriz ESC/ERS en 2022 [17]. Los valores objetivo anteriores se han modificado y ahora son los siguientes: mPAP (presión arterial pulmonar media) >20 mmHg, PAWP (presión arterial pulmonar en cuña) ≤15 mmHg, PVR (resistencia vascular pulmonar) ≥ 2 WU (unidades Wood*).
* unidad tradicional de medida de las resistencias de los vasos
” Utilizar “Ventanas de oportunidad
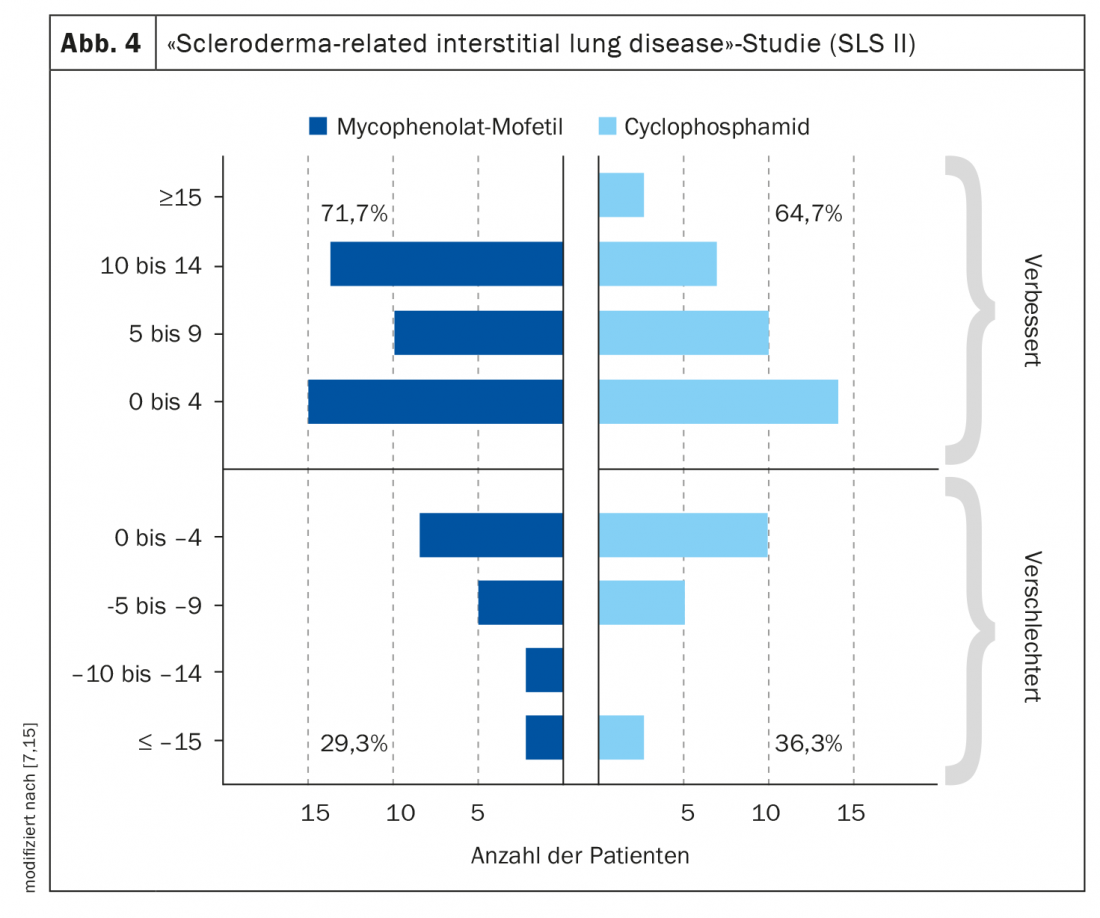
La distinción entre la SSc cutánea limitada y la SSc cutánea difusa sigue siendo relevante. Según las recomendaciones de la EULAR, el procedimiento terapéutico depende de la afectación del órgano [5]: 1. Síndrome de Raynaud, 2. úlceras digitales, 3. SSc-PAH, 4. manifestación cutánea y pulmonar, 5. Crisis renal, 6. manifestación gastrointestinal. Los dos fármacos más utilizados son la ciclofosfamida y el micofenolato mofetilo (MMF), basados en dos ensayos controlados aleatorizados con resultados de eficacia similares [6,7]. En el Estudio sobre la esclerodermia pulmonar (SLS) II, la ciclofosfamida y el MMF alcanzaron una eficacia comparable, pero el MMF presentó el mejor perfil de seguridad y tolerabilidad a largo plazo (Fig. 4). Por ello, el MMF se utiliza con mayor frecuencia en la práctica clínica. De gran interés, por supuesto, es la cuestión de qué implicaciones tiene un diagnóstico precoz para la terapia. En un estudio de cohortes retrospectivo publicado en 2022, los pacientes con SSc que manifestaron SSc cutánea difusa o enfermedad pulmonar intersticial en los 6 años siguientes al inicio de la enfermedad se dividieron en grupos de intervención precoz y tardía en función de la duración de la enfermedad. En los primeros, el tratamiento se inició en un plazo de ≤18 meses desde el inicio de la enfermedad, en los segundos sólo >18 meses después [4]. Las opciones de tratamiento farmacológico utilizadas fueron ciclofosfamida, MMF, metotrexato o tocilizumab. En el grupo de intervención temprana, la enfermedad activa disminuyó significativamente del 79% al 42% (p=0,007), mientras que el cambio en el grupo de intervención tardía no fue estadísticamente significativo (del 68% al 42%; p=0,11). En general, los hallazgos de este estudio apoyan la suposición de que existe una “ventana de oportunidad” para las opciones terapéuticas en los pacientes con SSc.
SSc-ILD: espectro terapéutico ampliado para la complicación potencialmente mortal
Para las manifestaciones cutáneas y pulmonares, la EULAR recomienda las siguientes opciones farmacológicas: Metotrexato, ciclofosfamida, trasplante autólogo de células madre hematopoyéticas, posiblemente MMF, posiblemente azatioprina. La enfermedad pulmonar intersticial (EPI) en la esclerosis sistémica (SSc-ILD) es actualmente la causa de muerte asociada a la enfermedad más común en pacientes con SSc [8]. La prevalencia de la enfermedad pulmonar intersticial como complicación de la SSc es de aproximadamente el 50% en una cohorte reciente basada en la población [9]. El principal tratamiento farmacológico utilizado hasta ahora han sido los inmunosupresores, siendo el MMF el fármaco más utilizado internacionalmente [10,11]. Estudios recientes sugieren que el nintedanib y el tocilizumab pueden ayudar a ralentizar el deterioro de la función pulmonar en la SSc-ILD [12]. El nintedanib es un inhibidor antifibrótico de la tirosina quinasa que se utiliza en la enfermedad pulmonar intersticial para prevenir la cicatrización del tejido pulmonar. En Suiza, el nintedanib (Ofev®) está aprobado desde 2020 para el tratamiento de la enfermedad pulmonar intersticial asociada a la esclerosis sistémica [12]. La dosis recomendada es de 150 mg dos veces al día a intervalos de unas 12 horas. El tocilizumab fue aprobado por la FDA estadounidense para el tratamiento de la SSc-ILD basándose en los datos de un ensayo de fase III [13].
La medicina de precisión señala el camino hacia el futuro
El Grupo de Trabajo de Terapia con Células Madre de la Sociedad Alemana de Reumatología publicó un documento de posición sobre el trasplante autólogo de células madre hematopoyéticas (ahSCT) para la esclerosis sistémica [14]. En consecuencia, la ahSZT es razonable en las siguientes condiciones: duración máxima de la enfermedad de 4 años, mRSS de min. 15, afectación de órganos internos u otros factores de pronóstico desfavorable, respuesta insuficiente a la ciclofosfamida o al MMF. La SSc es una enfermedad muy heterogénea. “Necesitamos una estratificación molecular a diferentes niveles OMICS a largo plazo”, afirma el Dr. Grasshoff [1]. Aquí es donde entra en juego la medicina de precisión. “El objetivo sería tratar la vasculopatía de forma vasoactiva, la inflamación de forma inmunomoduladora y la fibrosis de forma antifibrótica”, dijo [1]. Los estudios que correlacionan la firma génica intrínseca con la respuesta a diferentes fármacos son un enfoque prometedor para una estrategia de tratamiento dirigido específica para cada subgrupo con un perfil beneficio-riesgo optimizado.
Congreso: Congreso alemán de reumatología
Literatura:
- “Esclerosis sistémica: una enfermedad heterogénea”, Dra. Hanna Grasshoff, Congreso alemán de reumatología, 31.08.-03.09.2022.
- Minier T, et al: Ann Rheum Dis 2014; 73(12): 2087-2093.
- González García A, Callejas-Rubio JL: European Journal of Internal Medicine 97(Suppl 113); DOI:10.1016/j.ejim.2021.12.012
- Yomono K, Kuwana M: Reumatología (Oxford) 2022; 61(9): 3677-3685.
- Kowal-Bielecka O, et al: Ann Rheum Dis 2017; 76(8): 1327-1339.
- Tashkin DP, et al: NEJM 2006 (354): 2655-2666.
- Tashkin DP, et al: Lancet Respir Med 2016 (4): 708-719.
- Elhai M, et al: Ann Rheum Dis 2019; 78: 979-987.
- Hoffmann-Vold AM, et al: Am J Respir Crit Care Med 2019; 200: 1258-1266.
- Fernández-Codina A, et al: Arthritis Rheumatol 2018; 70: 1820-1828.
- Khanna D, et al: [abstract]. Arthritis Rheumatol 2018, 70 (suppl 9). https://acrabstracts.org/
- Información sobre medicamentos, www.swissmedicinfo.ch, (último acceso 07.11.2022)
- Khanna D, et al: Lancet Respir Med 2020; 8: 963-974
- Alexander T, Burmester G: Revista de Reumatología. Número 5/2020.
- Schneider U, et al: Z Rheumatol 2021; 80: 868-878.
- van den Hoogen F, et al: Arthritis Rheum 2013; 65(11): 2737-2347.
- Humbert M, et al: Grupo de documentos científicos ESC/ERS. EHJ 2022; 43(38): 3618-3731.
PRÁCTICA GP 2022; 17(11): 16-17