Thyroid nodules are a well-known phenomenon; fortunately, medullary thyroid carcinomas are not. It is a malignant degeneration of the C cells of the thyroid gland, which are located parafollicularly and cannot store iodine. Since it secretes calcitonin, this can be used as a tumor marker.
Thyroid nodules are frequently diagnosed in the office, but fortunately medullary thyroid carcinoma is rarely diagnosed. Only about 3% of all thyroid carcinomas involve medullary thyroid carcinoma (MTC), which has some special features: It is a malignant degeneration of the C-cells of the thyroid gland, which are parafollicular and cannot store iodine; it secretes calcitonin (Ctn) and CEA, which are used as tumor markers; a quarter of cases are familial in multiple endocrine neoplasia type 2 (MEN2). The only option for possible cure of MTC is early diagnosis and adequate surgery. This is made possible by consistent use of Ctn determination in the workup of struma nodosa and in the familial variant of RET protooncogene molecular genetic testing as part of family screening. Even in those not cured, MTC has a relatively favorable prognosis with a relatively good quality of life because of its slow growth. A risk-adapted “active surveillance” follow-up strategy is possible in many cases. MTC at the stage of symptomatic progressive distant metastasis can nowadays be treated with tyrosine kinase inhibitors (TKIs) [1,2].
Clinic and diagnostics of medullary thyroid carcinoma (MTC).
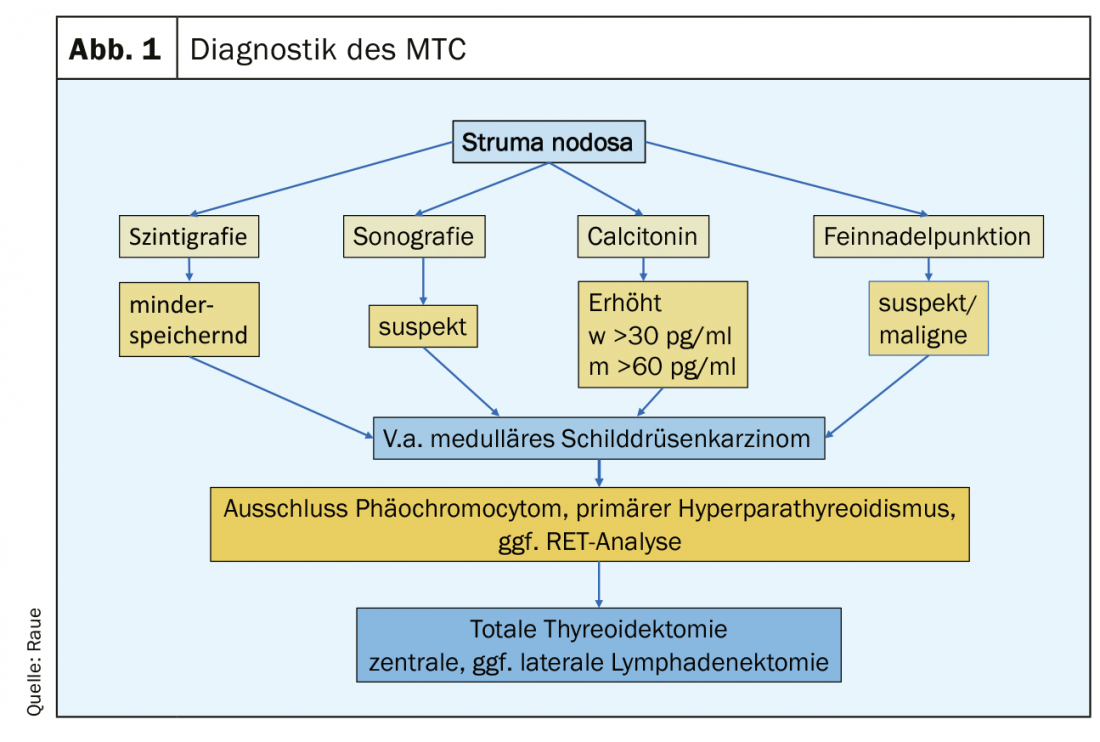
Clinically, medullary MTC does not differ significantly from other thyroid carcinomas: growing nodule in the thyroid gland, nonspecific neck discomfort, development of cervical lymph node swelling, and in the advanced metastatic stage, marked tumor-induced diarrhea may occur. Nowadays, MTC is diagnosed either as an incidental finding in the histology of a thyroid surgery specimen or on preoperative examination as part of the workup of a thyroid nodule (Fig.1). Indicative findings include a suspicious nodule on thyroid ultrasound (echo-poor, microcalcifications, fuzzy margins, extension deeper than wide), suspicious cervical lymph nodes, a suspicious fine-needle aspiration, and an elevated Ctn level. A Ctn level above 100 pg/ml is the most sensitive and specific finding. Affected are patients in the 4. and 5th decade of life. In younger patients, familial variant (MEN2) should be considered, which may occasionally manifest via concomitant conditions such as pheochromocytoma and primary hyperparathyroidism. The rare MTC should also be considered as part of the workup for CEA elevation.
Calcitonin screening in the workup of goiter nodosa
Ctn correlates with tumor mass and is a sensitive and specific tumor marker for early detection and follow-up of MTC. Routine determination of Ctns in the workup of goiter nodosa allows early diagnosis [3]. Slightly elevated Ctn levels are found in C-cell hyperplasia, which may be a precursor of hereditary MTC, but is also observed as a “benign” concomitant phenomenon in other thyroid diseases [4]. With development of newer sensitive and specific measuring fully automated Ctn assays (chemiluminescent immunoassays with sensitivity of 0.5 pg/ml and sex-specific upper reference values) [5,6], basal Ctn levels have become more important for screening in struma nodosa.
At preoperative Ctn levels above 100 pg/ml, MTC is present in nearly 100% of cases, whereas at Ctn levels between 10-20 pg/ml, the finding rate for MTC is less than 5% [7]. Taking into account sensitivity and specificity, gender-specific cut-off values of basal Ctn for recommending surgery for suspected MTC are approximately 30 pg/ml for women and approximately 60 pg/ml for men. The gray range of Ctn for women is 20-30 pg/ml, for men 30-60 pg/ml, in which 6-13% small MTC were missed [8]. Rising Ctn levels are more suggestive of MTC, in which case surgery should be recommended. Surgical measures can cure MTC in almost 100% up to a Ctn level of 100 pg/ml [9].
RET protooncogene – germline mutations in MEN2.
In approximately 25% of patients with MTC, a germline mutation in the RET protooncogene can be detected as evidence of MEN2 syndrome. Clinically, MEN2A with possible development of pheochromocytoma and hyperparathyroidism is distinguished from MEN2B with typical habitus, gangloneuromatosis of mucous membranes, and possible development of pheochromocytoma. Not all families can be captured by family history because families are often small or it is a late manifesting hMTC with only low penetrance. In a small percentage, a “de novo” mutation is observed in the patient; other family members are not affected. In a large series, a germline mutation in the RET protooncogene was found in 12% of apparently sporadic MTC. Therefore, molecular genetic testing for germline mutations in the RET gene should be initiated in all patients with an MTC.
In the hereditary form of MTC, the causative mutations are characterized in almost all families, mostly punctate mutations in 8 different exons of the RET protooncogene. Molecular genetic detection of these mutations, together with Ctn determination in families, allows early thyroidectomy with cure of MTC in affected children and adolescents. The optimal timing of thyroidectomy in RET mutation gene carriers is nowadays recommended based on the risk classification of the specific RET mutation into moderate, high and highest risk for early development of MTC (early or late penetrance) (Table 1) [1]. The Ctn level individually defines the prophylactic thyroidectomy to be planned early, so that in the optimal case an additional lymphadenectomy is not necessary.

Up to 50% of MEN2 patients develop pheochromocytomas during life, depending on the genotype, usually after manifestation of MTC. Pheochromocytomas may be multifocal and bilateral [10]. Patients with RET-634 and -918 mutations are particularly affected, and less frequently with mutations in exons 13-15. Up to 10% of MEN2 patients may develop primary hyperparathyroidism in a genotype-dependent manner; patients with a RET-918 mutation are not affected. Other extrathyroidal manifestations, such as ganglioneuromatosis in MEN2B or lichen amyloidosis interscapular, are also genotype dependent.
In tumor tissue of sporadic MTC, a somatic mutation in the RET gene, mostly RET-M918T, is frequently detectable, interestingly less frequently in smaller tumors, more frequently in larger ones, and most frequently in lymph node filiae and distant metastases, suggesting that this mutation is not the primary event in tumorigenesis. In tumor tissue in which no RET mutation is found, detection of a RAS mutation is often successful. Detection of the somatic RET mutation has prognostic significance and is a prerequisite for therapy with the selective RET tyrosine kinase inhibitors (LOXO-292, BLU-667) that are likely to become available soon [11].
Operation of the MTC
The primary therapy for MTC is surgical. Total thyroidectomy is the minimum procedure, supplemented by central and, if necessary, lateral unilateral/bilateral lymph node (LK) dissection, depending on the Ctn measured preoperatively and tumor stage. In case of an incidentally detected MTC in histology, the postoperative Ctn level should decide on the further procedure; if the Ctn is not measurably low, no further surgery is necessary; if the Ctn is elevated, depending on the tumor stage, a completion surgery (total TX and central and lateral LK dissection) should be performed. Surgical curative therapy is only possible if no distant metastases, no infiltration into soft tissues have been described in the primary histology and if less than 5-10 affected cervical LK are detectable in the previous histology or less than 3 affected compartments after systematic lymphadenectomy [12]. If this favorable situation is present, curative surgical therapy should be attempted. In the case of a hereditary MTC, total thyroidectomy is appropriate in every case, since in principle an MTC can develop from any C-cell. Of MTC diagnosed via a palpable thyroid nodule, 70% already have cervical lymph node metastases and 13% have distant metastases to liver, lung, or bone [13]. The 10-year survival is approximately 61-81% [14], depending on tumor stage at diagnosis, age, sex, pre- and postoperative Ctn levels. In the last decade, due to increasingly earlier diagnosis, more favorable tumor stages, and improved surgical strategies, survival has increased overall, but also in the higher tumor stages [14,15].
In patients with MEN2 detected by family screening, the timing of “prophylactic thyroidectomy” is based on RET mutation and Ctn level (cf. Tab. 1). Early diagnosis and complete surgery raised the disease-specific 5-year survival of all MTC to 89% [14].
Aftercare of the MTC
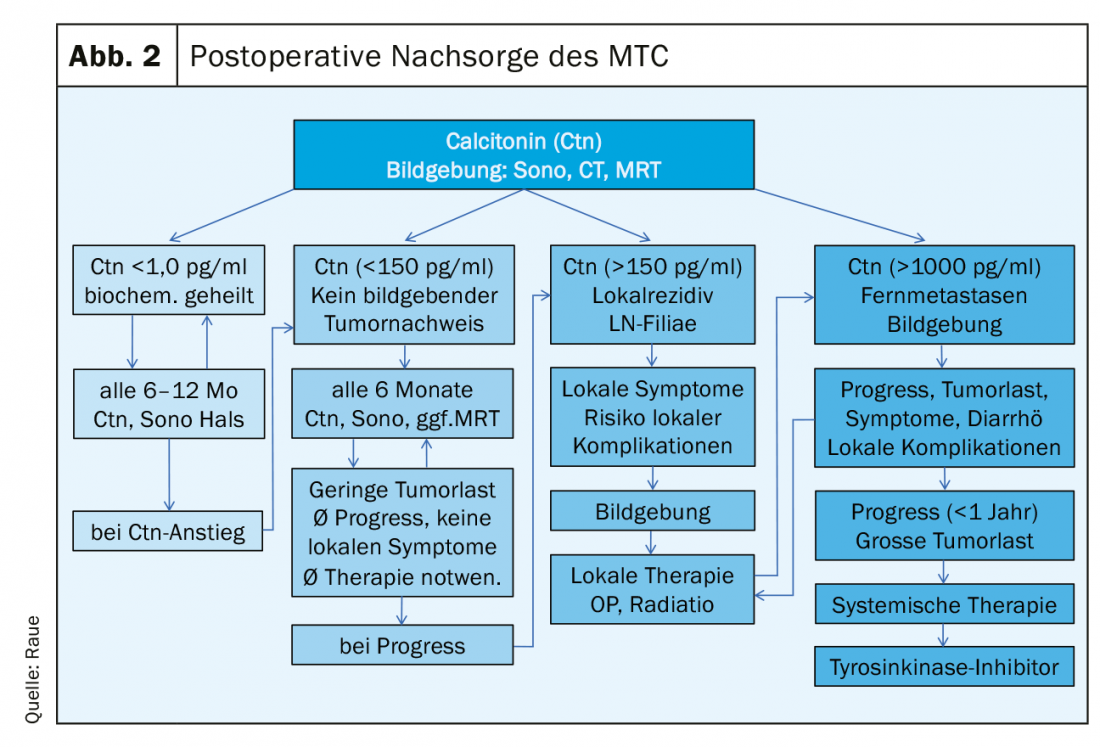
Postoperatively, the following results should be available to plan structured follow-up: the histology, Ctn immunohistology of the tumor if applicable, classification according to the pTNM scheme, the result of RET analysis for classification into the hereditary or sporadic variant, and the postoperative Ctn level. Three risk groups of patients can be defined in terms of response to primary therapy and prognosis: excellent (Ctn not measurably low), biochemically incomplete (Ctn measurable but no tumor tissue detectable by imaging), and structurally incomplete (Ctn markedly elevated, evidence of metastatic lymph nodes or distant metastases). (Fig. 2). In the further process, tumor marker(Ctn/CEA) doubling time calculation and imaging is performed to detect the possible tumor growth, then risk group adjustment is done [16,17]. Follow-up intervals are risk-adjusted and are performed every 3 months to once per year, depending on the size and location of the residual tumor/metastases and the extent of tumor progression [18]. Depending on the location of the suspected metastases, imaging procedures may include sonography of the neck and abdomen, computed tomography with contrast medium, MRI, bone scintigram, and possibly FDG-PET or F-DOPA-PET (Fig. 1).
Risk groups in follow-up care
If the response is excellent, the Ctn level is not measurably low: then the patient can be assumed to be biochemically cured, provided the histology is correct (Ctn immunohistology obligatory). In the first 2 years, semi-annual follow-up examinations with sonography of the neck, Ctn and CEA determination, review of substitution treatment with thyroxine (goal: TSH in the normal range) are sufficient, if necessary, in the case of postoperative hypoparathyroidism, calcium and calcitriol administration (goal: serum calcium in lower limit range 2.0-2.3 mmol/l) [19]. Thereafter, if no Ctn increase has occurred, it is possible to move to annual inspections. The prognosis is excellent and does not differ from the normal population.
In biochemically incomplete response, Ctn levels are persistently elevated usually low to moderate (usually below 1000 pg/ml). Remaining tumor tissue must be assumed. If the previous operation was not adequate, a “staging” is followed by a complementary adequate operation. However, once these limits (see section Surgery of MTC) are exceeded, a curative approach is no longer possible; therefore, all further therapeutic measures should be critically weighed, taking into account the risk of morbidity and the usually good quality of life. Often, despite intensive search, no definite tumor correlate is found with only moderately elevated Ctn up to 150 pg/ml. The progression of tumor disease as it progresses can vary widely and can be estimated relatively well using the Ctn and CEA doubling time [20]. Prerequisite for a good statement are at least 4 Ctn values over 2 years. Patients with tumor marker doubling times less than 24 months also had image morphologic progression in 94%. When tumor marker doubling times ran for 24 months, 86% of patients had no detectable tumor growth [21]. In the further course, semi-annual progress checks are usually sufficient. The frequency of diagnostic imaging can be planned based on primary findings, growth during progression (RECIST criteria), and tumor marker doubling time. A close-meshed maximum diagnosis without therapeutic consequence is not useful, e.g. sonography every 6 months, computed tomography/MRI every 12 months are often sufficient.
In structurally incomplete response, the Ctn level is significantly elevated (usually more than 1000 pg/ml): Here, local infiltrating tumor tissue, cervical lymph node metastases or distant metastasis mostly to lung, liver and/or bone must be assumed. A curative approach is no longer possible; palliative measures are in the foreground [22].
Re-operations of locoregional recurrence under a palliative approach are particularly useful in cases of progressive local recurrence or painful lymph node filiae in the central neck (trachea or esophagus proximity/infiltration) to reduce local complications. It is not reasonable to subject every newly detectable metastasis cervically to immediate surgery, as this leads to numerous non-target surgeries. There is no evidence that these surgeries positively affect the overall outcome; they are often associated with side effects such as recurrent paresis, hypoparathyroidism, and arm muscle paralysis. Radiation therapy is helpful for progressive unresectable local or mediastinal recurrences. Since surgery is more difficult after radiotherapy, operability should always be considered first from a palliative point of view.
Liver metastases are common, usually cause few symptoms, and are rarely an indication for surgery. Significantly progressive and painful liver metastases should be treated. In most cases, the metastasis is multiple and diffuse. Local therapy by (chemo)embolization or selective internal radiotherapy (SIRT) is of limited efficacy. Systemic therapy with tyrosine kinase inhibitors is useful in cases of rapid progression.
Bone metastases are rarely osteolytic, fracture prone, or painful. External radiotherapy is appropriate for painful bone metastases, and external radiotherapy or surgical intervention is appropriate for fracture risk. Bisphosphonate/denosumab therapy may be used in addition on an individual basis, especially in cases of pain or fracture risk, particularly in osteolytic metastases [23].
Chemotherapy is poorly effective in metastatic MTC. Tyrosine kinase inhibitors have been tested in trials for advanced metastatic MTC for about 10 years. Since May 2012, the tyrosine kinase inhibitor vandetanib has been available in Switzerland for the treatment of aggressive and symptomatic MTC; in some countries, including Germany, cabozantinib has also been approved for progressive MTC and has been indicated in this country for the treatment of advanced renal cell carcinoma since 2017. As evidence of benefit in early stage disease is clearly lacking, the current indication for therapy with TKIs is seen in high tumor burden and progressive (RECIST criteria, short tumor marker doubling time) and markedly symptomatic disease when local therapeutic measures have been exhausted. Prolongation of survival has not been demonstrated with any TKI in metastatic MTC to date [11].
Despite distant metastases, many patients have a good quality of life for years because tumor growth is often very slow. An “active surveillance” strategy is possible. The treatment of metastatic MTC is markedly symptom-oriented, including consistent antidiarrheal treatment in advanced tumor stages with loperamide and/or tinctura opii, which is often neglected or dosed too cautiously to the detriment of quality of life.
Follow-up care for MEN2
In addition to MTC follow-up, annual diagnostic testing regarding pheochromocytoma is performed starting at age 11 for high- and highest-risk RET mutations and at age 16 for moderate-risk RET mutations using metanephrine and catecholamine determination and MRI imaging, if necessary. With regard to primary hyperparathyroidism, annual control with serum calcium and parathyroid hormone determination is performed analogously with regard to age in the corresponding mutation risk groups (Tab. 1).
Take-Home Messages
- The tumor marker for medullary thyroid carcinoma (MTC) is calcitonin.
- Calcitonin screening in struma nodosa allows early diagnosis of MTC.
- All patients with MTC should have molecular genetic analysis of the RET protooncogene at diagnosis.
- Usually, the treatment of choice is total thyroidectomy with central lymphadenectomy.
- Follow-up differentiates between biochemically cured (Ctn not measurably low), biochemically incomplete (Ctn elevated without tumor evidence on imaging), and structural tumor evidence (metastases on imaging).
- In many cases with metastases, “active surveillance” is possible; in cases of progression and symptoms, the rule is: local (palliative surgery, radiation) before systemic (tyrosine kinase inhibitors).
Literature:
- Wells SA Jr, et al: Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015; 25(6): 567-610.
- Ceolin L, et al: Medullary thyroid carcinoma beyond surgery: advances, challenges, and perspectives. Endocr Relat Cancer 2019; 26(9): R499-R518.
- Raue F, Frank-Raue K: Calcitonin Screening in Nodular Goiter-Upper Limits. Dtsch Arztebl Int 2018; 115(13): 221.
- Costante G, et al: Determination of calcitonin levels in C-cell disease: clinical interest and potential pitfalls. Nat Clin Pract Endocrinol Metab 2009; 5(1): 35-44.
- Kratzsch J, Petzold A, Raue F, et al: Basal and stimulated calcitonin and procalcitonin by various assays in patients with and without medullary thyroid cancer. Clin Chem 2011; 57(3): 467-474.
- Kahaly GJ, Algeciras-Schimnich A, Davis TE, et al: United States and European Multicenter Prospective Study for the Analytical Performance and Clinical Validation of a Novel Sensitive Fully Automated Immunoassay for Calcitonin. Clin Chem 2017; 63(9): 1489-1496.
- Mian C, Perrino M, Colombo C, et al: Refining calcium test for the diagnosis of medullary thyroid cancer: cutoffs, procedures, and safety. J Clin Endocrinol Metab 2014; 99(5): 1656-1664.
- Frank-Raue K, Schott M, Raue F. On behalf of the thyroid section of the DGE. [Recommendation for Calcitonin Screening in Nodular Goiter]. Dtsch Med Wochenschr 2018; 143(15): 1065-1069.
- Machens A, Dralle H. Biomarker-based risk stratification for previously untreated medullary thyroid cancer. J Clin Endocrinol Metab 2010; 95(6): 2655-2663.
- Mucha L, Leidig-Bruckner G, Frank-Raue K, et al: Phaeochromocytoma in multiple endocrine neoplasia type 2: RET codon-specific penetrance and changes in management during the last four decades. Clin Endocrinol (Oxf) 2017; 87(4): 320-326.
- Cabanillas ME, Ryder M, Jimenez C: Targeted Therapy for Advanced Thyroid Cancer: Kinase Inhibitors and Beyond. Endocr Rev 2019; 40(6): 1573-1604.
- Machens A, Gimm O, Ukkat J, et al: Improved prediction of calcitonin normalization in medullary thyroid carcinoma patients by quantitative lymph node analysis. Cancer 2000; 88(8): 1909-1915.
- Roman S, Lin R, Sosa JA: Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer 2006; 107(9): 2134-2142.
- Randle RW, Balentine CJ, Leverson GE et al: Trends in the presentation, treatment, and survival of patients with medullary thyroid cancer over the past 30 years. Surgery 2017; 161(1): 137-146.
- Opsahl EM, Akslen LA, Schlichting E, et al: Trends in Diagnostics, Surgical Treatment, and Prognostic Factors for Outcomes in Medullary Thyroid Carcinoma in Norway: A Nationwide Population-Based Study. Eur Thyroid J 2019; 8(1): 31-40.
- Lindsey SC, Ganly I, Palmer F, et al: Response to initial therapy predicts clinical outcomes in medullary thyroid cancer. Thyroid 2015; 25(2): 242-249.
- Yang JH, Lindsey SC, Camacho CP, et al: Integration of a postoperative calcitonin measurement into an anatomical staging system improves initial risk stratification in medullary thyroid cancer. Clin Endocrinol (Oxf) 2015; 83(6): 938-942.
- Raue F, Frank-Raue K: Long-Term Follow-up in Medullary Thyroid Carcinoma. Recent Results Cancer Res 2015; 204: 207-225.
- Leidig-Bruckner G, Bruckner T, Raue F, et al: Long-Term Follow-Up and Treatment of Postoperative Permanent Hypoparathyroidism in Patients with Medullary Thyroid Carcinoma: Differences in Complete and Partial Disease. Horm Metab Res 2016.
- www.thyroid.org/professionals/calculators/thyroid-cancer-carcinoma
- Laure Giraudet A, Al Ghulzan A, Auperin A, et al: Progression of medullary thyroid carcinoma: assessment with calcitonin and carcinoembryonic antigen doubling times. Eur J Endocrinol 2008; 158(2): 239-246.
- Hadoux J, Pacini F, Tuttle RM, et al: Management of advanced medullary thyroid cancer. Lancet Diabetes Endocrinol 2016; 4(1): 64-71.
- Vogel T, Wendler J, Frank-Raue K, et al: Bone Metastases in Medullary Thyroid Carcinoma: High Morbidity and Poor Prognosis Associated With Osteolytic Morphology. J Clin Endocrinol Metab 2020; 105(6).
FAMILY PRACTICE 2020, 15(7): 14-18









