When one or both chambers of the heart are inappropriately thickened or dilated, the heart is no longer as efficient. Cardiomyopathies are characterized by a variety of different causes of such changes in the heart muscle tissue. However, the differentiation of each clinical manifestation has major implications for prognosis and potential treatment regimens.
Cardiomyopathies have many faces. Basically, they can be classified into four morphological phenotypes with hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), arrhythmogenic cardiomyopathy (ARVC), and restrictive cardiomyopathy (RCM). However, this rough, view-based classification has rather little in common with the actual causes, can be illustrated by the large number of subcategories. Prof. Benjamin Meder, MD, Heidelberg (D), showed that imaging alone is therefore not sufficient to detect the disease. Accordingly, the functional characteristics should still be added. This is because cardiomyopathy is ultimately the description of a morphological and functional myocardial phenotype that cannot be explained by coronary artery disease or altered filling properties due to arterial hypertension, valvular vitiation, or congenital heart disease.
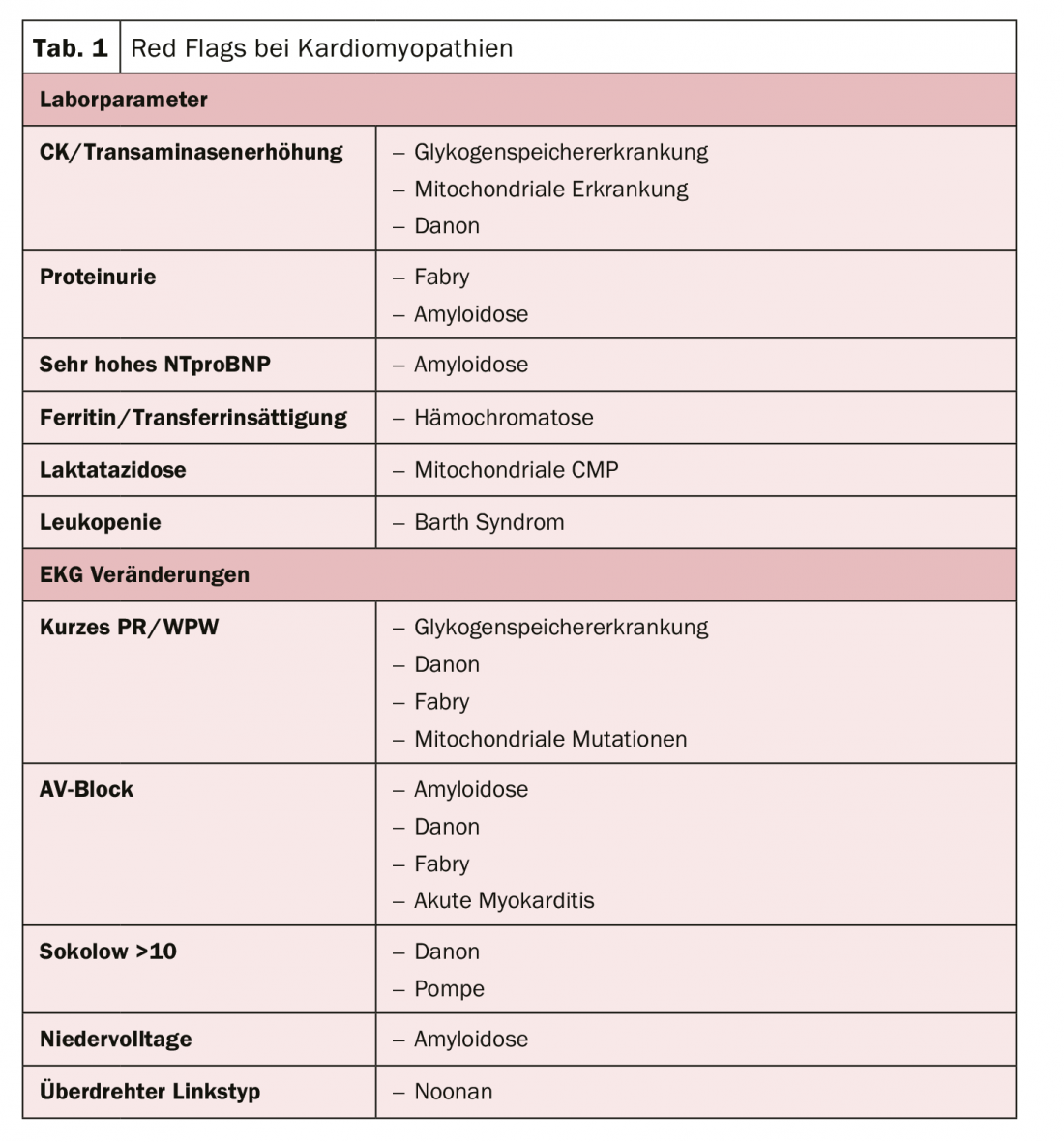
The structural approach to optimized treatment therefore approaches via etiology with the goal of avoiding sudden cardiac death, the limiting problem of all cardiomyopathies. There are typical laboratory parameters and abnormalities in the ECG that can indicate the presence of cardiomyopathy (tab. 1). Being able to make these distinctions is therefore relevant, as the prognosis is essentially determined by the cause, says the expert. In particular, patients with amyloidosis have very poor survival.
Detect and effectively treat amyloidosis
Amyloidosis can be divided into three types based on organ involvement: AL amyloidosis affecting the kidneys, heart and/or intestines, mt-ATTR amyloidosis affecting mainly the heart or nervous system, and wt-ATTR amyloidosis affecting mainly the heart. In principle, AL amyloidosis can be understood as a complication of plasma cell dyscrasia. Therefore, hematologic therapy along the lines of that used in multiple myeloma is used with the goal of eliminating the amyloidogenic, (cardio)toxic light chains as quickly as possible. In addition to high-dose chemotherapy for fit patients, a proteasome inhibitor-based regimen is primarily used. In younger patients, CyBorD (bortezomib, cyclophosphamide, dexamethasone) should be favored over BMDex (bortezomib, melphalan, dexamethasone) because of the stem cell toxicity of melphalan to preserve the option of subsequent stem cell apheresis and high-dose chemotherapy.
In amyloidosis caused by transthyretin protein deposition, drugs such as diflunisal and tafamidis can stabilize the mutant protein. Furthermore, gene therapies that reduce transthyretin production (e.g., patisiran, inotersen) may reduce nervous system effects.
For patients with symptomatic cardiac AL and ATTR amyloidosis, the same general therapy recommendations apply in principle as for heart failure patients. However, even low doses of β-blockers or ACE inhibitors can lead to symptomatic hypotension. Therefore, treatment in patients with cardiac amyloidosis is primarily based on proper dosing of diuretics.
Syndromal involvement of many organ systems – Fabry disease
Fabry disease is a genetically inherited disorder that usually manifests between the ages of 20-40. A misfolded or nonfunctional α-Gal A enzyme is formed, disrupting transport from the endoplasmic reticulum to the lysosome. This leads to an accumulation of lysosomal substrates. Besides the heart, the kidneys, the central nervous system, but also the peripheral nervous system and the skin can be involved. If not treated, renal failure is the most common cause of death in these patients, explained Prof. Ingrid Kindermann, MD, Homburg/Saar (D). Cardiac symptoms occur in more than half of all Fabry patients. Malignant arrhythmias are mainly responsible for sudden cardiac death.
For a long time, therapy was limited to the mere alleviation of symptoms. There is now a specific treatment option in which the missing enzyme is replaced by a biotechnologically produced enzyme. Laboratory-produced alpha-galactosidase A is administered to the patient by infusion and ensures that the accumulated storage material is broken down. However, since enzyme replacement therapy cannot repair organ damage that has already occurred, but only delay its progression, it should be used as early as possible. Another option is to take a pharmacological chaperone orally. This agent can be used in patients with certain mutations in the GLA gene and residual activity of the α-galactosidase A enzyme. This is because it binds to unstable forms of AGAL and stabilizes the enzyme so that it can break down the accumulated lipids in the cell.
Source: CardioMedLive 2020
CARDIOVASC 2020; 19(3): 28-29 (published 9/17/20, ahead of print).