The collective term “pulmonary hypertension (PH)” encompasses several life-threatening types of pulmonary hypertension whose therapeutic approaches vary widely and are highly complex. Since the symptoms are often unspecific, even the diagnosis is a great challenge.
The clinical classification of pulmonary hypertension (PH) is currently divided into five groups, as Prof. Dr. Horst Olschewski, Head of the Clinical Division of Pulmonology, University Department of Internal Medicine, LKH University Hospital Graz, reminded the audience at the outset:
- Group 1 includes Pulmonary Arterial Hypertension (PAH), the rarest form of pulmonary hypertension, which is diagnosed only after other underlying causes have been ruled out.
- Group 2, pulmonary hypertension in left heart disease, is by far the most common form of pulmonary hypertension.
- Group 3 includes pulmonary hypertension associated with pulmonary disease and/or oxygen deprivation. This form is also much more common than PAH.
- Group 4 includes pulmonary hypertension after pulmonary embolism, which affects up to 4% of patients who have suffered an acute pulmonary embolism.
- Group 5 includes rare forms of pulmonary hypertension that cannot be clearly assigned to any of the previously mentioned groups, such as pulmonary hypertension in sarcoidosis or chronic kidney disease.
Groups 2 and 3, Prof. Olschewski said, together account for a total of 90% of all pulmonary hypertension, which poses differential diagnostic hazards.
Heterogeneity of the IPAH
Patients who met the criteria for idiopathic pulmonary arterial hypertension (IPAH) were identified for cluster analysis.
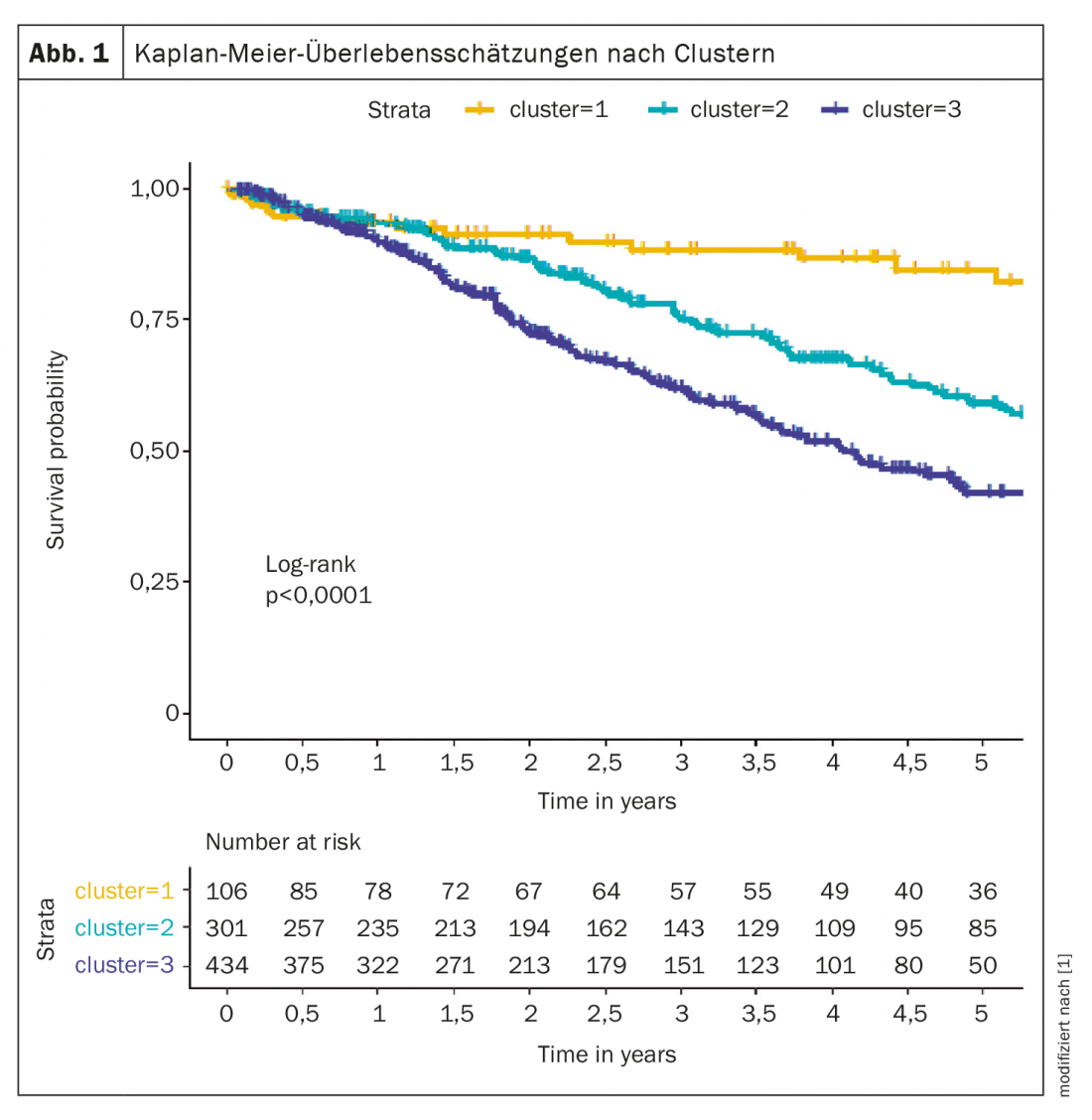
For example, cluster analysis identified several phenotypes that differed in clinical presentation, response to therapy, and survival. The analyzed groups were composed from the COMPERA registry [1], three clusters were defined: cluster 1 (n=106; 12.6%) included a mean age of 45 years, 76% were women, no comorbidities, mostly never smokers, DLCO ≥45%; cluster 2 (n=301; 35.8%) included patients whose mean age was 75 years, 98% women, frequent comorbidities, no smoking history, DLCO mostly ≥45%; cluster 3 (n=434; 51.6%) consisted of subjects with a mean age of 72 years, 72% were men, common comorbidities, smoking history, and DLCO mostly below 45%.
Patients in cluster 1 responded better to PAH treatment than patients in the other two clusters. Five-year survival was 84.6% in cluster 1, 59.2% in cluster 2, and 42.2% in cluster 3 (Fig. 1). According to the expert, these data suggest that criteria are necessary to distinguish atypical IPAH patients from true IPAH patients.
Differential diagnosis sometimes difficult
Because therapeutic consequences depend largely on the cause of pulmonary hypertension, it is important to complete diagnostic procedures and determine the primary cause of PH before making a decision about PAH medications. The World Symposia on Pulmonary Hypertension (WSPH) has produced guidelines for these important decisions. PH in the 2 group or complex developmental diseases with elevated postcapillary pressure are relatively easy to identify by elevated pulmonary arterial wedge pressures. Group PH 4 can be detected or excluded by perfusion lung scans in combination with chest CT. Group 1 PAH and Group 3 PH are quite different disease profiles but can sometimes be difficult to distinguish. WSPH suggests that severe pulmonary hypertension in combination with mild impairment on pulmonary function test (FEV1 >60 and FVC >60%), mild parenchymal abnormalities on high-resolution CT of the chest, and circulatory impairment on cardiopulmonary exercise test are indicative of group PAH 1. These patients are candidates for PAH therapy. If the patient suffers from group 3 PH, the only possible indication for PAH therapy is severe pulmonary hypertension (mPAP ≥35 mmHg or mPAP between 25 and 35 mmHg along with a very low cardiac index (CI) <2.0 L/min/m2), which can only be derived invasively, Prof. Olschewski said.
One study also addressed the differentiation between patients with PAH (group 1) and patients with heart failure (group 2) [2]. Although increased left-sided filling pressures and functional mitral regurgitation primarily lead to postcapillary PH, guidelines and recommendations differentiate between isolated postcapillary PH (IpcPH) and combined post- and precapillary PH (CpcPH). The latter is defined by pulmonary vascular resistance (PVR) elevated to Wood units (WE). It is important to differentiate between the general definition of PH (mPAP >20 mmHg) and the definition of precapillary PH including PAH, for which a pulmonary arterial wedge pressure (PAWP) ≤15 mmHg and an increase in pulmonary vascular resistance (PVR) to ≥3 Wood units are also required. According to this study, targeted drug therapy for PAH is indicated when PAWP is ≤15 mmHg.
The differentiation of patients with PAH and COPD (group 1) from patients with PH due to COPD (group 3) is also not straightforward. Most COPD patients with PH are in group 3. Some patients with COPD with PH and increased left ventricular filling pressure (postcapillary PH) caused by concomitant cardiovascular disease are assigned to group 2. Chronic thromboembolic disease can also cause PH, especially because COPD is a risk factor for venous thromboembolism, these patients with COPD are usually assigned to group 4. Patients with very mild peripheral airway obstruction and severe precapillary PH with markedly increased pulmonary vascular resistance (PVR) and low cardiac output (CO) are predominantly thought to have PAH (group 1) with mild COPD as concomitant disease. In most cases, PH is relatively mild in COPD patients, the pulmonologist explained, but in a subset of COPD patients, the presence of certain clinical features suggests a “pulmonary vascular phenotype.” Such a phenotype would be characterized by severe precapillary PH with markedly increased pulmonary vascular resistance, moderate airflow limitation, severely reduced diffusing capacity for carbon monoxide, normo- or hypocapnia, circulatory load limitation, and progressive right heart failure.
Another study attempted to determine prognostically relevant hemodynamic thresholds for severe PH in COPD using an unbiased approach [3]. Here, a PVR >5 WU proved to be the strongest independent hemodynamic predictor of mortality in COPD patients. This threshold is most appropriate for identifying COPD patients with severe pulmonary vascular disease.
Advances in drug therapy
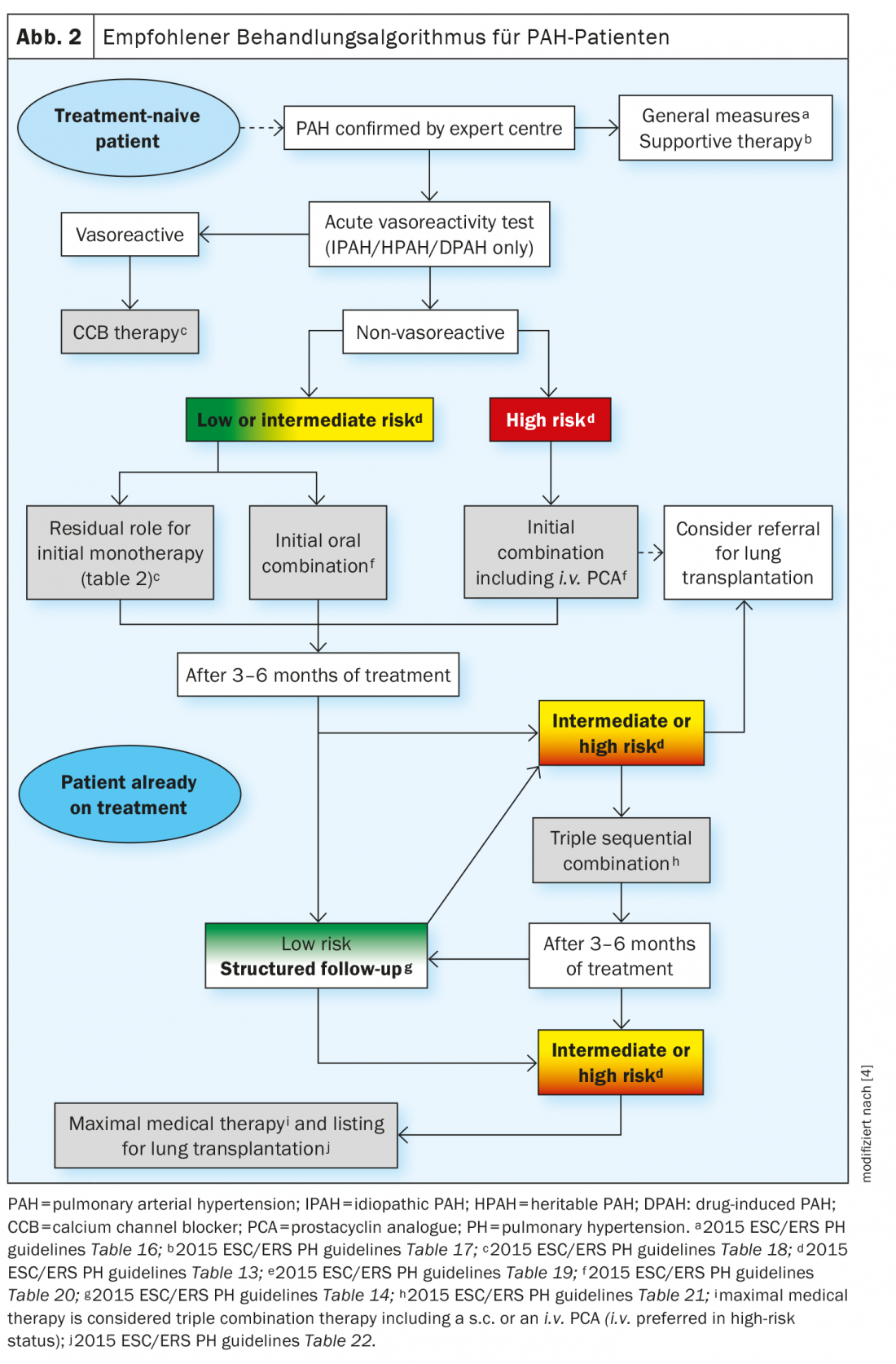
Recent advances in drug therapy for PAH have not resulted from the discovery of new signaling pathways, but rather from the development of new strategies for combination therapy and escalation of treatments based on systematic assessment of clinical response. The treatment strategy is based on the severity of the newly diagnosed PAH patient, which is determined using a multiparametric risk stratification approach. Clinical, exercise, right ventricular function, and hemodynamic parameters are combined to define low, intermediate, or high risk status according to expected 1-year mortality. The current treatment algorithm provides the most appropriate initial strategy, including monotherapy, dual or triple combination therapy. Further escalation of treatment is required if low-risk status is not achieved at scheduled follow-up visits. In most advanced cases, lung transplantation may be required with maximal medical therapy (Fig. 2).
Approved oral therapy medications include phosphodiesterase 5 inhibitors (PDE5i) such as sildenafil and tadalafil, endothelin receptor antagonists (ERA) such as bosentan, armbrisentan, and macitentan, soluble guanylate cyclase (sGC) stimulators such as riociguat, and prostacyclin receptor agonists such as selexipag. In addition, PAH can be treated with inhaled therapy or continuous infusions.
Prospective studies on therapy
For example, riociguat and phosphodiesterase-5 inhibitors (PDE5i), which are approved for the treatment of pulmonary arterial hypertension (PAH), act through different mechanisms in the same pathway. The REPLACE trial therefore investigated whether riociguat could be an alternative option for patients with PAH who do not respond adequately to treatment with PDE5i. The aim here was to evaluate the effects of switching to riociguat from PDE5i therapy compared with continued PDE5i therapy in patients with PAH at intermediate risk of 1-year mortality. The results indicate that switching from treatment with PDE5i to riociguat, both of which act through the signaling pathway between nitric oxide-soluble guanylate cyclase and cyclic guanosine monophosphate, may be a strategic option for treatment escalation in patients with PAH at intermediate risk of 1-year mortality [5].
Sotatercept, a novel activin antagonist, binds activins and growth differentiation factors in an attempt to restore the balance between growth-promoting and growth-inhibiting signaling pathways. In the PULSAR trial, 24 weeks of treatment with sotatercept resulted in a reduction in pulmonary vascular resistance in patients with pulmonary arterial hypertension who were receiving background therapy for it [6]. Physical performance (measured by 6-minute walk distance) and NT-proBNP levels also improved with sotatercept, Prof. Olschewski said.
“Sensation” from the USA
Prof. Olschewski had a “sensation” to report from the USA: There has never been a therapy approved for PH in lung diseases (group 3). That has now changed – at least in the USA. Inhaled treprostinil was used there for the first time in this group of patients. The 16-week placebo-controlled INCREASE trial enrolled sufferers with interstitial lung disease and pulmonary hypertension who were administered inhaled treprostinil via an ultrasound nebulizer with pulsed administration in up to 12 breaths (72 μg total) four times daily. Compared with placebo, inhaled treprostinil significantly improved patients’ physical performance as determined by 6-minute walk test [7]. In the USA, the active ingredient has already been approved by the FDA, but whether the manufacturer will also apply to the European Medicines Agency (EMA) in the near future or wait for further studies is still open, as the expert explained. “In any case, though, it’s a glimmer of hope.”

Standardized movement training successfully established
The EU-TRAIN randomized controlled trial of exercise training in patients with pulmonary arterial hypertension (PAH) and chronic thromboembolic pulmonary hypertension (CTEPH), conducted in 11 centers in 10 European countries in a large patient population, demonstrated significant and clinically meaningful improvement in the primary endpoint of 6MGT and secondary endpoints of WHO-FC, QoL, and maximal oxygen consumption [8]. The study demonstrated for the first time that a safe and effective exercise program can be standardized as an adjunct to drug therapy and is feasible in different countries with different health care systems.
Source: Pneumo Update 2021: Pulmonary Hypertension, 12.11.2021
Literature:
- Hoeper, et al: Idiopathic pulmonary arterial hypertension phenotypes determined by cluster analysis from the COMPERA registry. J Heart Lung Transplant 2020, doi: 10.1016/j.healun.2020.09.011.
- Rosenkranz, et al: Pulmonary hypertension in HFpEF and HFrEF: Pathophysiology, diagnosis, treatment approaches. Heart 2019, doi: 10.1007/s00059-019-4831-6.
- Zeder, et al: Elevated pulmonary vascular resistance predicts mortality in COPD patients. Eur Respir J 2021, doi: 10.1183/13993003.00944-2021.
- Galiè, et al: Risk stratification and medical therapy of pulmonary arterial hypertension. European Respiratory Journal 2019, doi: 10.1183/13993003.01889-2018.
- Hoeper, et al: Switching to riociguat versus maintenance therapy with phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension (REPLACE): a multicentre, open-label, randomised controlled trial. Lancet Respir Med 2021, doi: 10.1016/S2213-2600(20)30532-4.
- Humbert, et al: Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2021, doi: 10.1056/NEJMoa2024277.
- Waxman, et al: Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med 2021, doi: 10.1056/NEJMoa2008470.
- Grüning, et al: Standardized exercise training is feasible, safe, and effective in pulmonary arterial and chronic thromboembolic pulmonary hypertension: results from a large European multicentre randomized controlled trial. Eur Heart J 2021, doi: 10.1093/eurheartj/ehaa696.
InFo PNEUMOLOGY & ALLEROLOGY 2022; 4(1): 20-22.