
This very rare syndrome is caused by mutations in the LRP2 gene, which codes for megalin, and is inherited in an autosomal recessive manner. It is characterized by craniofacial features with characteristic facial features. In addition to ocular complications and sensorineural hearing loss, other clinical abnormalities are described. The suspected diagnosis can be confirmed by genetic testing. Depending on the individual course, various therapeutic measures are required.
Donnai-Barrow syndrome (DBS) is associated with multiple congenital malformations [1]. Affected individuals are characterized by typical facial dysmorphism, myopia and other ocular findings, as well as hearing loss, agenesis of the corpus callosum, low molecular weight proteinuria and various intellectual developmental disorders. Congenital diaphragmatic hernia and/or omphalocele are common. Little data is available on the prevalence and incidence of DBS. To date, fewer than 50 affected individuals from around 20 families have been described. DBS occurs in all ethnic groups and there does not appear to be a gender gradient. DBS is caused by mutations in the LRP2 gene (low-density lipoprotein receptor-related protein 2; 2q31.1). This codes for megalin, which is expressed in several resorbing epithelia – particularly in the brain, kidneys and eyes. Megalin plays an important role in various signaling cascades and in the endocytosis of many ligands.
Common clinical features
Almost all patients exhibit the following symptoms [1]:
- Agenesis/hypogenesis of the corpus callosum
- Enlarged anterior fontanel
- pronounced sensorineural hearing loss (i.e. the sound reaches the inner ear but either cannot be converted into nerve impulses or these are not transmitted to the brain)
- Hypertelorism.
Characteristic facial features are [1]:
- downward sloping palpebral fissures,
- Short nose with a flat nasal bridge,
- High broad forehead
- “widow’s peak” in the front hairline and sometimes proptosis
About 40% of patients have a congenital diaphragmatic hernia and/or omphalocele. Development is often delayed and intelligence is reduced to varying degrees. High myopia (>6 dptr) can lead to retinal detachment/dystrophy and increasing visual impairment. Occasionally, iris coloboma, focal segmental glomerulosclerosis and proximal tubule dysfunction (rarely leading to renal insufficiency) have been reported.
| Diagnosis and DD The diagnosis of Donnai-Barrow syndrome (DBS) is made by a combination of clinical and imaging features together with a typical pattern of low molecular weight proteinuria, elevated urinary levels of retinol-binding protein (RBP) and RBP/creatinine ratio. The diagnosis is confirmed by DNA analysis. Due to the characteristic constellation of symptoms, the number of differential diagnoses of DBS is limited. Some common symptoms include tetrasomy 12p, as well as Fryns, Chudley-McCullough, acrocallosal and cranio-fronto-nasal syndromes. The renal phenotype is partly similar to Dent disease and Lowe syndrome. The ocular phenotype may indicate Stickler syndrome. The detection of hypertelorism and congenital diaphragmatic hernia or omphalocele in prenatal imaging must suggest DBS. Prenatal diagnosis in high-risk pregnancies requires prior identification of the disease-causing mutations in the family. |
| according to [1] |
Therapeutic management and prognosis
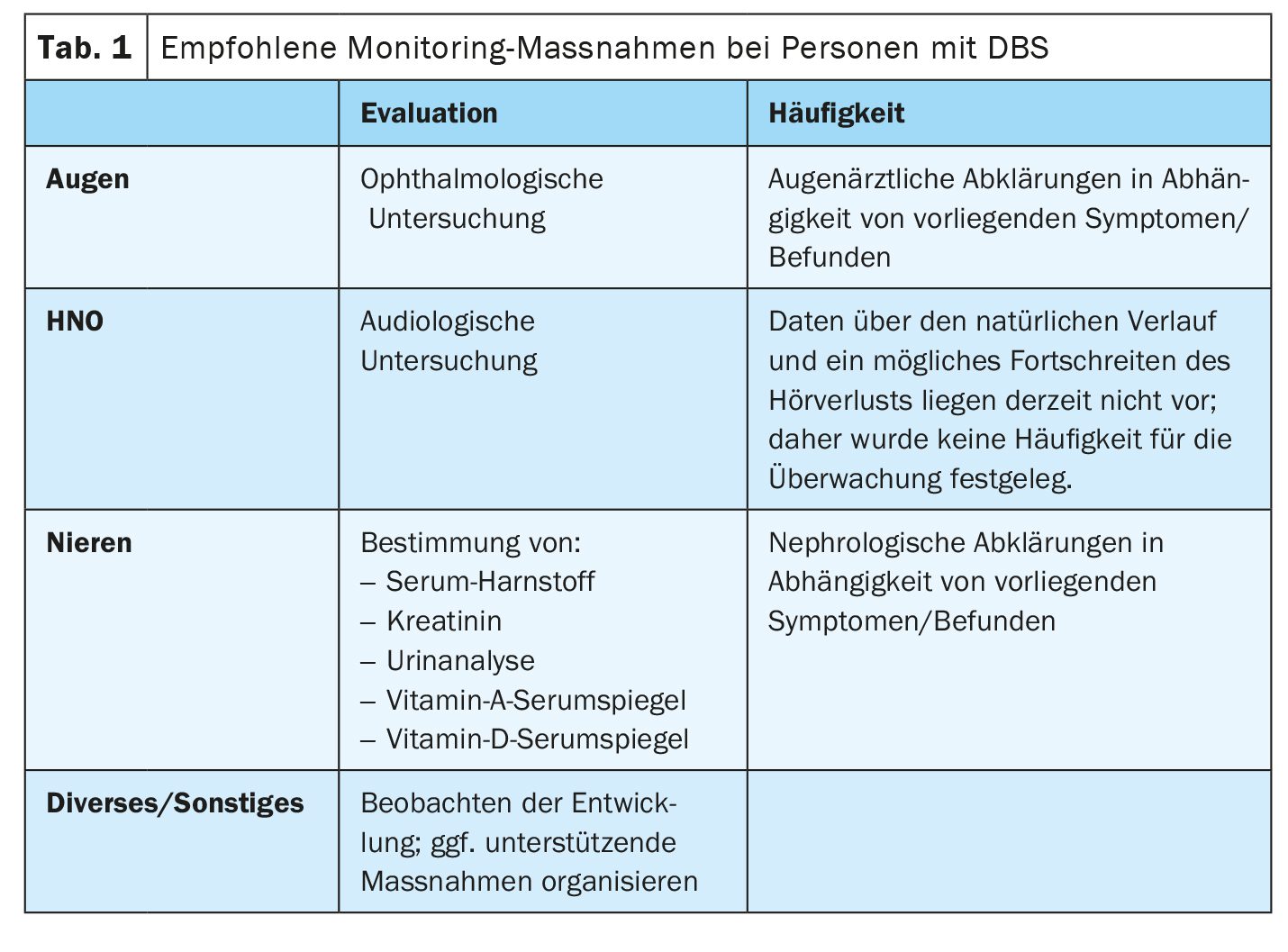
DBS is inherited in an autosomal recessive manner. The parents of affected children and their relatives should be offered genetic counseling [1]. With the exception of a single published case of uniparental disomy, the parents of patients whose cases were documented were found to be obligate heterozygotes. Regular check-ups of visual acuity, hearing and kidney function are necessary. Spectacle correction, treatment to prevent retinal detachment, hearing aids and/or cochlear implants are part of the treatment plan. Congenital diaphragmatic hernia and/or omphalocele may require surgical intervention. Specific support measures should be offered for affected children. Those affected can achieve usable vision and hearing with correction. The general state of health of patients in childhood and adolescence is generally good. End-stage renal failure is a rare and life-threatening complication. Pre- or perinatal presentation with diaphragmatic and abdominal wall defects requires surgical intervention and is associated with increased morbidity and mortality.
Case study: Progression from birth to primary school age
A currently 9-year-old boy was born to healthy, unrelated Caucasian parents who were 34 years old (mother) and 40 years old (father) at the time [2]. He had a healthy sister and two healthy half-siblings on his mother’s side. During the pregnancy, a small exomphalos was detected by ultrasound. The patient was delivered by normal vaginal delivery. Postnatal clinical and imaging examinations revealed pronounced hypertelorism, bilateral coloboma (cleft formation in the eye area), absence of the corpus callosum, malrotation of the intestine, bilateral inguinal hernias, but no congenital diaphragmatic hernia.
Omphalocele: The umbilical hernia was reduced on the first day, his hernias were repaired at 1 year of age and the malrotation was operated on definitively at 18 months of age.
Corpus callosum agenesis: At the age of 4 months, the patient’s head circumference was 44.5 cm (95th percentile), height 60.6 cm (90th percentile) and weight 5.78 kg (90th percentile). The MRI scan of the brain confirmed the corpus callosum agenesis and also showed a frontal encephalocele with a widened anterior fossa and a Chiari 1 malformation with cerebellar tonsils extending to C1.
Ocular manifestations: In addition to bilateral iris and chorioretinal colobomas, high myopia was present, as well as a right inferior cataract and left posterior lenticonus, which had been diagnosed at 3 months of age. The myopia was associated with enlarged eyeballs (axial length of 30 mm at 7 years of age) and bilateral posterior staphylomata, and he underwent prophylactic 360-degree laser retinopexy to prevent retinal detachment. His spectacle prescription was OD (Oculus Dexter) -15.00 D, OS (Oculus Sinister) -19.25/-2.00 axis 92°, although he normally wore contact lenses and achieved a corrected visual acuity of OD 20/200, OS 20/100. At the age of 7 years, the patient had normal electrodiagnostic results, but reported visual deterioration over the past 2 years, especially at night. A repeat electroretinography at the age of 9 years revealed generalized retinal dysfunction affecting both the rod and cone systems, mainly at the interface between the photoreceptor and the retinal pigment epithelium. The ocular masses at 6 years of age were 45, 70 and 110 mm for the inner canthal, pupillary and outer canthal distance, respectively.
Audiological findings: Due to severe bilateral deafness, the patient was fitted with cochlear implants at the age of 4 years, which were revised at the age of 6 years and again at the age of 8 years.
School development: The patient attends a mainstream school and receives special support for his visual and hearing deficits. He has a certain developmental delay and attends a class in which he is two years behind his peers. However, he is making good progress in this class and it is thought that much of his developmental delay is due to his bisensory impairment and the gaps in his schooling due to frequent hospitalizations.
Diagnostic confirmation: The suspected diagnosis of DBS was confirmed by genetic testing, in which a homozygous 4-bp deletion (c.11469_11472delTTTG) in exon 60 of the LRP2 gene was detected by direct sequencing.
Literature:
- “Donnai-Barrow syndrome”, www.orpha.net,(last accessed 27.09.2024).
- Kantarci S, et al: Donnai-Barrow syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy. Am J Med Genet A 2008; 146A(14): 1842-1847.
- Longoni M, et al: Donnai-Barrow Syndrome. 2008 Aug 28 [Updated 2018 Nov 21]. In: Adam MP, et al. (Eds). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
FAMILY PHYSICIAN PRACTICE 2024; 19(10): 24-25









