
Fanconi-Bickel syndrome (FBS) occurs due to variants in the SLC2A2 gene. The diagnosis of a rare genetic disease can take up to 5-6 years, even longer in low and middle income countries with limited technological resources. Doctors from Peru presented the case of a two-and-a-half-year-old child with failure to thrive, hepatomegaly, metabolic acidosis, hypophosphatemia, hypokalemia and hyperlactatemia.
Fanconi-Bickel syndrome (OMIM #227810), an autosomal recessive (AR) inherited disorder, is characterized by a combination of liver and kidney disease caused by a defect in the glucose transporter GLUT2 (SLC2A2 gene). This leads to glycogen accumulation, proximal renal tubular dysfunction and impaired glucose and galactose utilization.
The phenotype includes lack of weight gain, abdominal distension, hepatomegaly, fasting hypoglycemia, postprandial hyperglycemia, glucosuria, phosphaturia, aminoaciduria, polyuria, metabolic acidosis, osteoporosis, hypophosphatemia, rickets, and the presence of glycogen in liver or kidney biopsy. In rare cases, hepatocellular carcinoma has been observed due to activation of the Wnt signaling pathway. However, cases of patients with mild clinical symptoms have also been reported, including those with pure glucosuria. The SLC2A2 gene (OMIM *138160) contains 11 exons, and its GLUT2 protein consists of 524 amino acids and is located in the cell membrane, expressed in hepatocytes, enterocytes, renal proximal tubules, pancreatic beta cells, neurons and astrocytes. Pathogenic variants of SLC2A2 alter glucose entry and exit in hepatocytes and reduce insulin secretion due to increased sensitivity of beta cells in the postprandial phase.
Case report of a 2½ -year-old boy
A boy aged 2 years and 7 months, born and raised in Peru from the fifth pregnancy of related parents, presented to the team of Dr. Hernán Abarca-Barriga of the Instituto Nacional de Salud (INS) Peru due to vomiting, diarrhea, metabolic acidosis, hypokalemia and hyperlactatemia, hypoactivity and fever. Due to these symptoms, he had previously been hospitalized at the age of 1 year and 10 months and 2 years and 2 months [1].
The child was born with a birth weight of 3620 g, a height of 49 cm and a head circumference of 34 cm (normal percentile) and an Apgar score of 8-9. In terms of psychomotor development, he achieved head control at one month, sat unaided at seven months, walked with support at one year and six months. He spoke his first words at one year and five months, said two words at two years and nine months and showed a social smile at one year. At 1 year and 8 months of age, he was evaluated for underweight and reduced growth, chronic diarrhea, fever, and increased abdominal volume (Fig. 1).
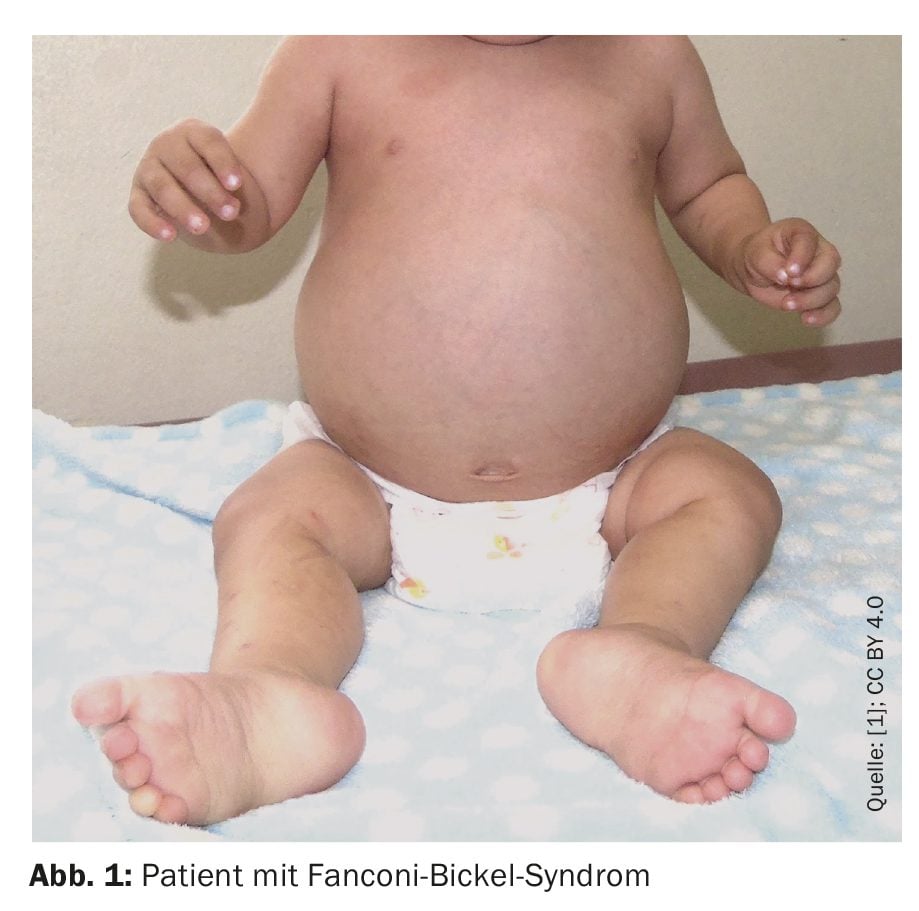
On admission, the doctors found a bulging forehead, hepatomegaly, hypotonia and a pseudo-madelung deformity. The child’s weight and height had been below the first percentile since he was six months old, while his head circumference was within normal limits. Radiographs showed fraying and widening of the metaphyses of the femur (distal) and tibiae (proximal), consistent with rickets.
The clinical and laboratory findings were suggestive of FBS
The small patient had hypoglycemia, hypo- and hypercalcemia, hypophosphatemia, hypercholesterolemia, hypertriglyceridemia, hyperphosphatasemia, hypokalemia with diarrhea and vomiting, hyperlactatemia, and hypouricemia. Urinalysis showed normal pH and HCO3 values, but hyperproteinuria, hypocreatinuria, microalbuminuria, hyperglycosuria and hypercalciuria. In addition, the boy had thrombocytosis. The venous gas showed a pH of 7.261-7.5 mmHg, HCO3 of 7.5-28.8 mmHg and a base excess of -15.8 to +5.3. These analyses led to the diagnosis of renal tubular acidosis. Abdominal ultrasonography showed hepatomegaly, there were no signs of fibrosis or nephromegaly.
A liver biopsy revealed a partially distorted liver architecture due to the presence of some fibrous enlargement of the portal space, inflammatory infiltration of lymphocytes, large and ballooned hepatocytes with a mosaic pattern and mild pericellular fibrosis. Periodic acid-Schiff staining with diastasis highlights eosinophilic deposits in hepatocytes correlating with glycogen deposition. Based on these clinical and laboratory findings, the physicians suspected Fanconi-Bickel syndrome (FBS).
Exome sequencing identified homozygous pathogenic variant
Genetic test results were obtained at 1 year and 8 months of age using genomic DNA. A total of 131,477 annotated variants were identified in 18,179 genes, excluding variants that were likely to be benign or mild. To identify variants associated with the patient’s phenotype, the terms “failure to thrive” (HPO: 0001508) and “hepatomegaly” (HPO: 0002240) were used, and the authors note that a population allele frequency threshold of 1% was considered. Due to hypophosphatemia, rickets and glycogen storage in the liver, the researchers also manually searched for variants in SLC2A2. Due to parental consanguinity, priority was given to the analysis of homozygotes. A variant allele frequency (VAF) of more than 0.9 was used to select potential candidate genes. Exome sequencing identified a homozygous nonsense variant in the gene SLC2A2, which was described as pathogenic.
The patient’s clinical diagnosis was based on the presence of hepatomegaly, hypoglycemia, glucosuria, hypophosphatemia, hyperphosphatasia, hypouricemia, rickets, pseudo-madelung deformity and renal tubular acidosis. However, the presence of aminoaciduria could not be determined as the test was not available on site. This inadequate excretion of amino acids would have facilitated the clinical biochemical diagnosis, the authors explain. Molecular confirmation using Sanger sequencing of the SLCA2 gene was another obstacle, as this test is not available in Peru. Given the limited access to genetic testing in the country, it was not possible to confirm the presence of the variant in the patient’s first-degree relatives, the authors emphasize. Exome sequencing enabled the researchers to accurately identify the homozygous variant in this case.
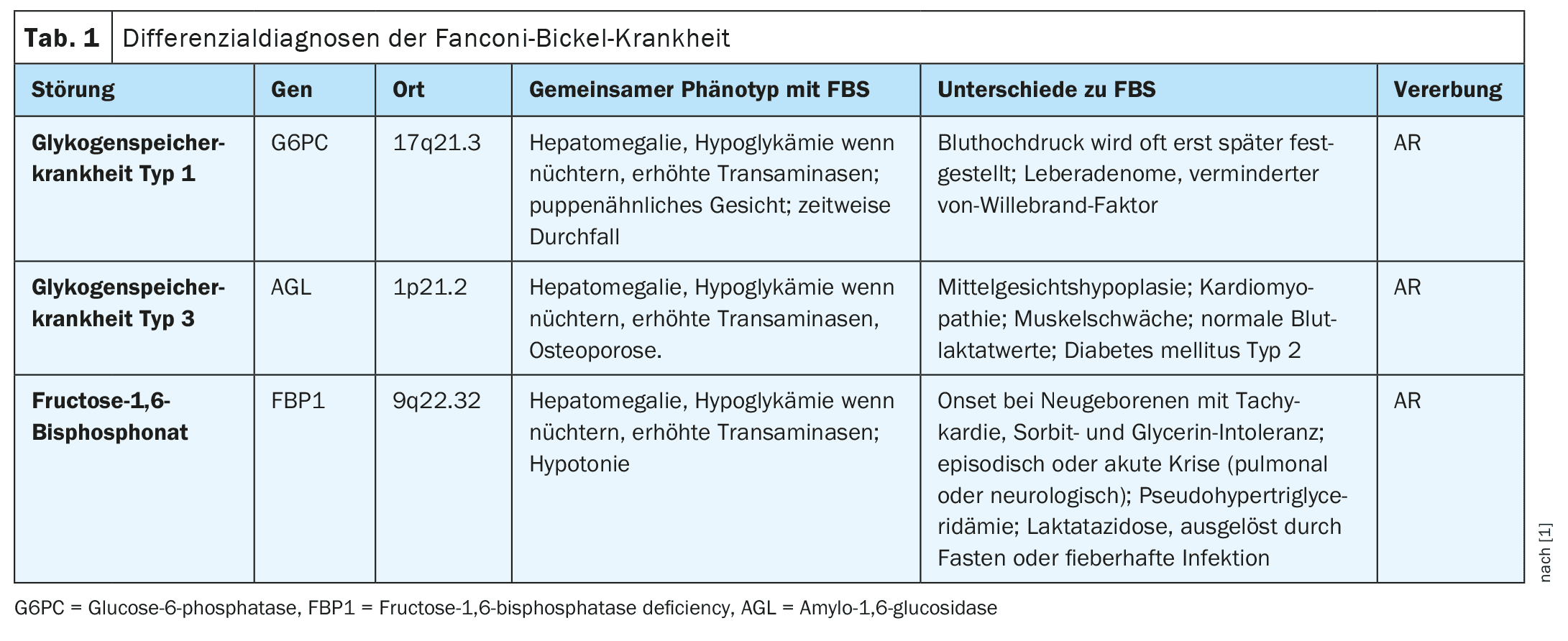
According to Dr. Abarca-Barriga and colleagues, the observed increase in blood lactate could be due to an increased glucose load from anaerobic metabolism. In addition, a low uric acid level due to proximal tubule dysfunction is part of the phenotype in FBS patients, leading to hyperuricosuria. Some clinical features, such as delayed speech development, are also likely to be associated with the presence of chronic hypoglycemia.
There is currently no causative therapy for Fanconi-Bickel syndrome. The young patient was treated with bicarbonate, amlodipine, sodium citrate and citric acid solution, enalapril, alendronate and zolendronate and received dietary treatment with uncooked cornstarch, which led to an improvement in weight and height. In addition, the authors emphasize that the patient showed an improvement of 1 standard deviation (SD) in weight and height since starting dietary treatment with uncooked corn starch. Therefore, the use of cornstarch is essential not only to prevent nocturnal hypoglycemia, but also to improve the patient’s height and weight.
Literature:
- Abarca-Barriga HH, et al.: Importance about use of high-throughput sequencing in pediatric: case report of a patient with Fanconi-Bickel syndrome. BMC Pediatr 2024; 24: 161; doi: 10.1186/s12887-024-04641-1.
HAUSARZT PRAXIS 2024; 19(11): 48–49









