Congenital heart defects are the most common organ malformation in children. Every cardiac defect requires pediatric cardiology evaluation and classification. Specialized pediatric cardiology follow-up is essential throughout childhood and adolescence in view of the child’s growth, possible residual cardiac findings after intervention/surgery, and any late complications.
Congenital heart defects are the most common organ malformation, occurring in about 1% of all newborns. For Switzerland, this corresponded to approximately 850 children out of 85 287 live births in 2014. They range from simple cardiac defects that have little effect on the cardiovascular system to severe ones (Table 1) that can lead to early death if left untreated [1].

The frequency with which the individual heart defects occur varies greatly (Tab. 2) and each heart defect requires a pediatric cardiological evaluation and classification. The most important diagnostic examination method here – in addition to the clinical examination including. Auscultation – echocardiography, which allows visualization and definition of virtually all cardiac structures.

The electrocardiogram (ECG) (also 24h Holter ECG) detects any arrhythmias or conduction abnormalities as well as hypertrophy signs. Depending on the situation, further special examination methods such as spiroergometry, cardiac magnetic resonance imaging (cMRI), computed tomography (CT) or cardiac catheterization for invasive diagnostics (e.g. pressure measurement, angiographies) are indicated. The latter also offers the possibility of intervention at the same time. Thus, most heart defects today are diagnosed in the first year of life. Without surgical or interventional therapy, life expectancy is significantly shortened in moderate and severe heart defects.
The extraordinary advances in cardiac surgery, interventional cardiology, anesthesia and intensive care medicine achieved over the last decades have significantly improved the chances of survival after cardiac surgery in childhood. Thus, more than 90% of children with congenital heart defects nowadays reach adulthood [2,3]. However, they often face multifaceted problems during their lifetime due to their cardiac pathophysiology and the concomitant diseases that are often also present. A key point is that most cardiac defects can be repaired but not corrected in the true sense [4].
Significant residual cardiac findings and fetal growth make specialized pediatric cardiology follow-up essential. Optimal follow-up of children with heart disease requires close collaboration of primary care physicians and office-based pediatricians with the involved specialists. This is particularly important in the case of complex concomitant diseases that affect neurological development, for example [5].
In adolescence, the transition to adult medicine to a specifically trained cardiologist (GUCH cardiologist; GUCH = Grown-Ups with Congenital Heart Disease) must be initiated in time in terms of a continuous flow of information [6].
Complications in the course
Arrhythmias: Some heart defects are associated with early arrhythmias. For example, complete atrioventricular block may occur with cc transposition of the great arteries. Most commonly, however, arrhythmias occur because of residual hemodynamic effects, such as sustained pressure loading with ventricular fibrosis or volume loading and/or myocardial scarring (atrio-ventriculotomy), after cardiac surgery. A rhythm disorder, incl. Conduction disorder, may develop early or late postoperatively. Observed are supraventricular tachycardia (usually reentry tachycardia), atrial flutter or fibrillation, ectopic atrial tachycardia, sick sinus syndrome, ventricular tachycardia, atrioventricular block, and sudden cardiac death.
While the risk of arrhythmia remains low after simpler cardiac surgeries, it increases significantly after complex procedures such as Fontan palliation in univentricular hearts. Therefore, (long-term) ECG and ergometry are part of the regular medical evaluation of these patients. For arrhythmias occurring late postoperatively, invasive electrophysiology is available today in addition to drug therapy, which achieves good results by means of ablation of e.g. intracardiac scars.
Physical limitations: With the increasing number of children reaching adulthood after surgical reparation or palliation, the inclusion of children in the social fabric with their peers has become an important goal. While a heart-healthy patient can increase his cardiac output fivefold during exercise, a patient with a complex heart defect, such as after Fontan palliation in the univentricular heart, is at best able to double it. This results in limited physical resilience [7].
Sports activities in the endurance area are generally supported. In contrast, isometric weight training or competitive sports are discouraged in most patients.
Testing and assessment of physical performance by spiroergometry is a regular part of pediatric cardiological follow-up from the age of about ten years.
Failure to thrive/neurocognitive development: Children with severe heart defects can develop failure to thrive and are then smaller and lankier in stature than healthy children of the same age. Diverse studies have shown that children with complex heart defects are at increased risk for neurocognitive deficits after cardiac surgery in neonatal or infancy. Delayed development of fine and gross motor skills, behavioral problems, reduced attention, hyperactivity symptoms, and language delay are common deficits [6,8]. This can have a negative impact on everyday life and educational and professional careers in adulthood. Children with mild to moderate heart defects are mostly unaffected. It is important to raise these issues with parents and offer targeted support with the supervising pediatrician or family physician.
Endocarditis: Infective endocarditis is a serious and potentially life-threatening complication in patients with congenital heart disease or acquired heart disease. Therefore, parent and patient education on the occasion of pediatric cardiological control is of great importance. Symptoms of endocarditis should be recognized early so that an adequate response can be made. Likewise, the benefits as well as correct implementation of drug endocarditis prophylaxis must be communicated. Current guidelines for endocarditis prophylaxis in children in Switzerland are listed in Table 3.

Pulmonary arterial hypertension: all congenital heart defects with large intra- or extracardiac shunts result in unrestrained volume and pressure loading of the pulmonary vascular bed. As a result, pulmonary arterial hypertension may develop, leading to irreversible changes in the late stages of this serious disease. For example, in the case of a large uncorrected ventricular septal defect with left-to-right shunt, there is an increase in pressure in the pulmonary vascular bed over years, which ends in shunt reversal with cyanosis due to new right-to-left shunt (Eisenmenger reaction). One of the aims of surgical intervention in early childhood is to prevent this development. Patients with congenital heart disease have an increased risk of developing pulmonary arterial hypertension even without the presence of relevant shunts. Depending on its severity, this has a detrimental effect on quality and duration of life [10]. For diagnosis and initiation of a specific therapy, a connection to a specialized center is usually required.
Concomitant diseases: Congenital heart defects are associated with other malformations and/or chromosomal abnormalities in 15-20%. Children with trisomy 21 have vitium cordis in 40-50% of cases. These patients, with often complex malformations, benefit from multidisciplinary care.
Examples of follow-up and specific aspects of individual heart defects
Aortic isthmus stenosis (AIST): the goal is to eliminate the stenosis early and create an aorta that is as gradient-free and normal-caliber as possible. This can only be achieved by surgery or interventional cardiac catheterization (balloon dilatation or stent implantation) (Fig. 1). The method of treatment depends on the age of the patient as well as the morphology of the AIST. Associated malformations often exist (e.g., bicuspid aortic valve in approximately 50% of cases).
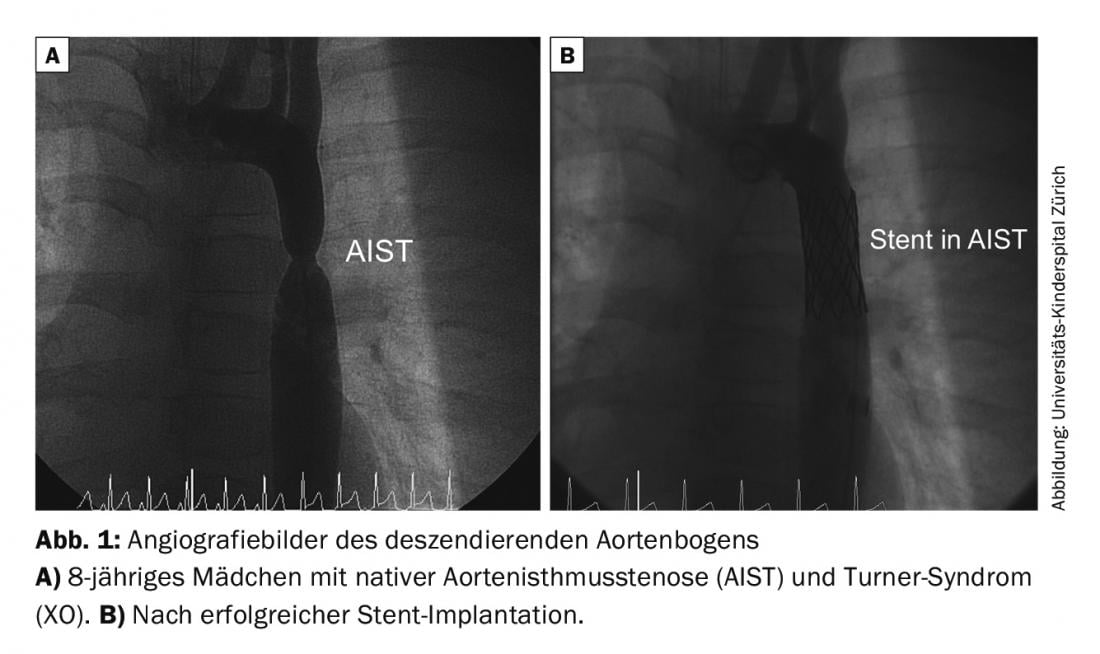
Patients with surgical repair have a 5-10% risk of re-stenting during follow-up. The risk of arterial hypertension increases with age, even if the stenosis has been effectively removed. Other potential risks include the development of aortic aneurysms/dissections at the suture site or stent.
Follow-up recommendations [11]:
- usually annually, in the course of min. every 2 years:
- clinical status
- Blood pressure measurement (BP) on all 4 extremities
- depending on resting BP: 24h BP measurement
- ECG
- Echocardiography (left ventricular size, myocardial thickness and function, degree of stenosis over aortic arch/sthmus, pay closer attention to associated malformations, if any).
- Ergometry (from 10 years) every 3-4 years
- cMRI: in case of insufficient visualization in echocardiography: visualization of the aortic arch, exclusion of aneurysm formation every 5 years.
d-transposition of the great arteries (d-TGA): Nowadays, arterial switch surgery to restore ventriculo-arterial concordance of d-TGA is the treatment method of first choice. The aorta and the pulmonary artery are interchanged and by the so-called Lecompte maneuver the pulmonary bifurcation is finally located in front of the aorta (Fig. 2) . At the same time, reimplantation of the coronary arteries is necessary. The anatomic pulmonary valve thus serves as a neo-aortic valve in the systemic circulation for life.
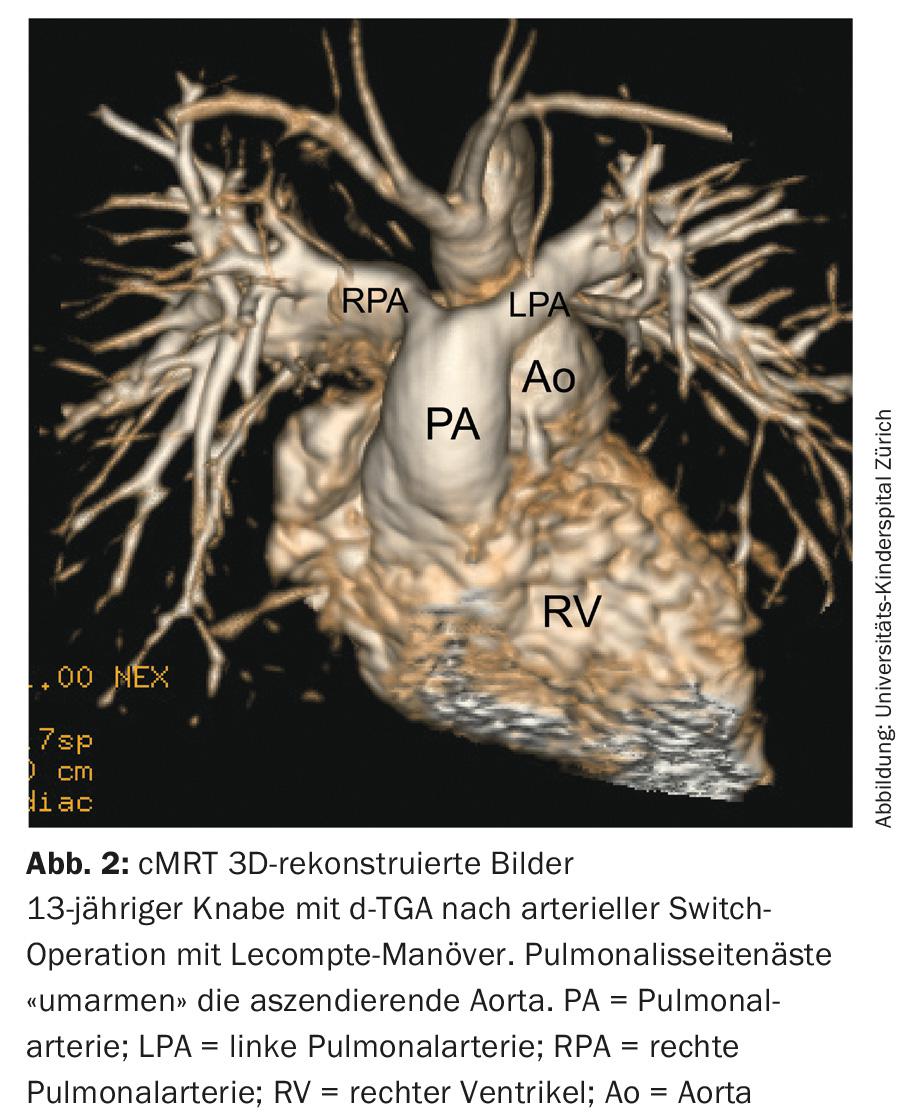
In the long term, dysfunction of this neo-aortic valve and dilatation of the aortic root may occur. Due to the lecompte maneuver with displacement of the pulmonary collateral branches anterior to the ascending aorta, torsion and stretching may occur, resulting in stenosis of the pulmonary collateral branches. Since the surgery involves coronary transfer, torsion and stretching can also cause stenosis or, in extreme cases, occlusion of a coronary artery, with consecutive myocardial infarction and sudden cardiac death.
Follow-up recommendations [9,11]:
- usually annually, in the course of min. every 2 years:
- clinical status
- ECG
- Echocardiography (ventricular size and function, degree of stenosis pulmonary side branches, wall mobility disorder, function neo-aortic valve, dimension of neo-aortic root)
- Long-term ECG according to individual assessment
- Coronary angiography in complicated perioperative course, symptomatic patient or abnormal routine diagnosis.
- Ergometry (from 10 years) every 2-3 years
- cMRI: in the presence of poor sound quality, for evaluation of right and left ventricular size and function, and assessment of pulmonary side branches. Coronary artery assessment and/or myocardial scarring.
Tetralogy of Fallot (TOF): the goal in anatomic surgical repair of tetralogy of Fallot is VSD patch closure and elimination of right ventricular outflow tract obstruction (RVOTO). If the pulmonary valve annulus is small in diameter and/or the valve is dysplastic, implantation of a transannular patch is necessary, which always leads to pulmonary valve insufficiency (Fig 3) . In the medium to long term, this results in relevant right ventricular dilatation, which can lead to ventricular dysfunction, impaired performance, and arrhythmia and ultimately necessitates pulmonary valve replacement.

Complete right bundle branch block is common after VSD patch closure; the risk of complete atrioventricular block necessitating pacemaker implantation is small (<2%).
Follow-up recommendations [9,11]:
- usually annually
- clinical status
- ECG (QRS width)
- Echocardiography (residual RVOTO including pulmonary side branches, pulmonary valve regurgitation, right ventricular size and function, residual VSD, left ventricular size and function, aortic root dimension).
- Long-term ECG (non-sustained tachycardia) min. every 3 years in asymptomatic patients
- Ergometry (from 10 years), every 3-5 years
- cMRI: with increasing RV dilatation: for evaluation of right ventricular size and function and assessment of pulmonary regurgitation fraction, left ventricular size and function. Assessment of pulmonary side branches.
Single Ventricle (SV): In univentricular hearts, a basic distinction is made between two basic forms, depending on whether the right or left ventricle has not sufficiently formed (e.g., tricuspid atresia or hypoplastic left heart syndrome; Fig. 4). Surgical therapy in the form of usually three staged palliative interventions ends in “Fontan circulation”. The goal is to create a series-connected circuit from the parallel circuits that have existed up to now. In the neonatal period, stabilization of the parallel circulation is needed first, which usually involves shunt placement or reduction of pulmonary hyperperfusion by banding, depending on the cardiac defect. In a second step, a bidirectional upper cavopulmonary (Glenn) anastomosis is created in infancy, resulting in significant volume decompression of the heart. With Fontan completion at 2-3 years of age using extracardiac conduit, total cavopulmonary anastomosis, definitive palliation is achieved in patients with univentricular hearts. Only with this final circulatory separation is cyanosis (except for small residual shunts; collaterals or fenestration) abolished.
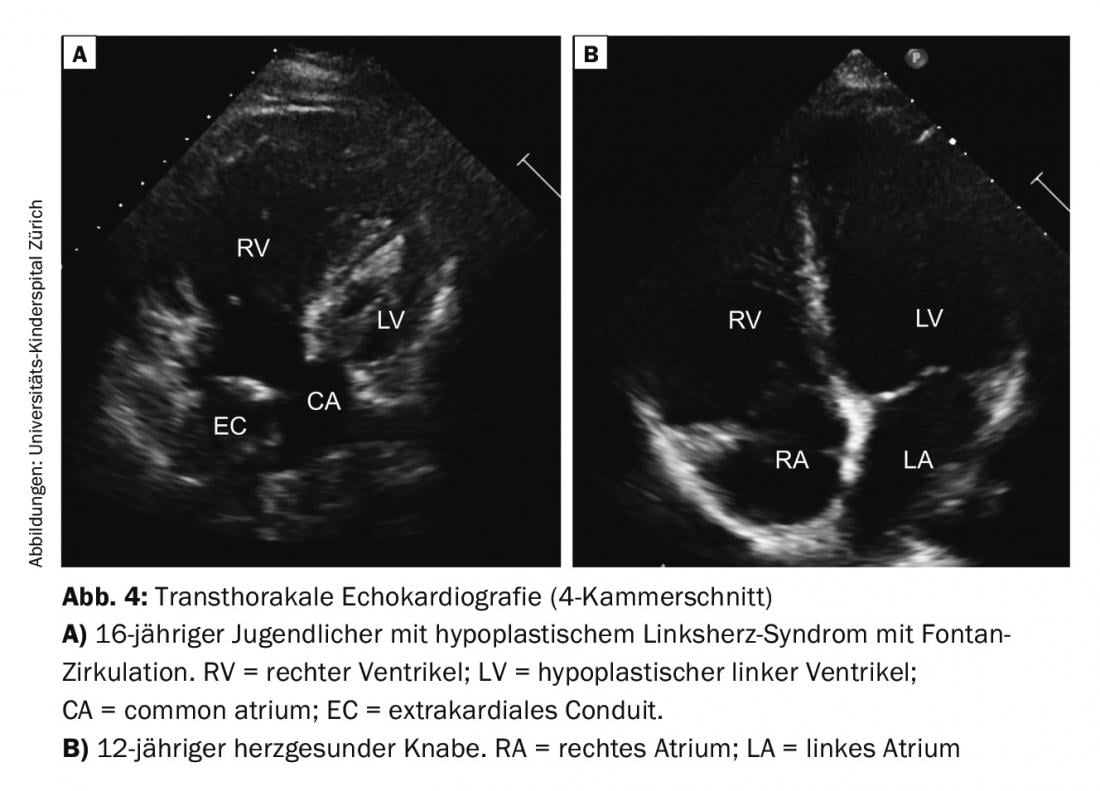
The care during the individual surgical steps and follow-up of this heterogeneous and unique group of patients with all their complications and late effects is a great challenge for the pediatric cardiologists and later for the specialized adult cardiologists (GUCH cardiologist).
The complications in these patients are many: reduced ventricular function, AV valve regurgitation, stenosis in the systemic or pulmonary circulation, arrhythmia, chronotropic incompetence, thrombosis/embolism, protein-losing enteropathy, plastic bronchitis, hepatic dysfunction, pulmonary hypertension, diaphragmatic paresis, scoliosis, etc.
Recommendations for follow-up after fontan completion [9,11]:
- min. annual
- clinical status
- BP measurement on all 4 extremities
- Transcutaneous oxygen saturation
- ECG
- Echocardiography (SV function, AV valve insufficiency, left obstruction (subaortic stenosis, isthmic stenosis), flow profile of upper cavopulmonary anastomosis or in Fontan circulation and hepatic veins, etc.).
- Laboratory examination: total protein or serum albumin, liver values, pro-BNP as cardiac function parameter, hemoglobin.
- Long-term ECG (nonsustained intraatrial re-entrant tachycardia, chronotropic competence), if there is evidence of arrhythmias or at least. all
- 3 years
- Ergometry (from 10 years), min. Every 3 years
- cMRI: assessment of ventricular function, quantification of blood flow, anatomical visualization of Fontan connections. Performed depending on clinical course, but usually recommended in teens and before transition to GUCH cardiologists.
- Diagnostic cardiac catheterization: in case of worsening Fontan circulation such as decrease in exercise capacity, increasing cyanosis, ascites or protein-losing enteropathy, plastic bronchitis, etc.
Closing words
Nowadays, the majority of congenital heart defects have a good long-term prognosis. Early detection of the heart defect, surgical and/or interventional treatment by an experienced team, and careful follow-up care make a decisive contribution. Close collaboration between the pediatrician and pediatric cardiologist, as well as other specialists if necessary, and later transition to a GUCH cardiologist is essential.
Literature:
- Hoffmann JIE, et al: The Incidence of Congenital Heart Disease. JACC 2002; 39(12): 1890-1900.
- Warnes CA, et al: Task Force 1: The Changing Profile of Congenital Heart Disease in Adult Life. JACC 2001; 37(5): 1161-1198.
- Moons P, et al: Temporal Trends in Survival to Adulthood Among Patients Born with Congenital Heart Disease from 1970 to 1991 in Belgium. Circulation 2010; 122: 2264-2272.
- Oechslin E: Congenital heart defects: lifelong specialized care for a chronic disease. Cardiovascular Medicine 2006; 9: 373-375.
- Mackie A: Children and Adults with Congenital Heart Disease Lost to Follow-Up. Circulation 2009; 120(4): 302-309.
- Bauersfeld U: Transition, transfer and cooperation in patients with congenital heart defects – continuous collaboration of pediatric and adult cardiology. Cardiovascular Medicine 2006; 9: 336-341.
- Gewillig M, et al: Failure of the Fontan Circulation. Heart Failure Clin 2014; 10(1): 105-116.
- Sterken C, et al: Neurocognitive Development After Pediatric Heart Surgery. Pediatrics 2016; 137(6), doi: 10.1542/peds.2015-4675.
- Wernovsky G, et al: Guidelines for the Outpatient Management of Complex Congenital Heart Disease. Congenit Heart Dis 2006; 1: 10-26.
- Dimopoulos K, et al: Pulmonary hypertension related to congenital heart disease: a call for action. Eur Heart J 2014; 35(11): 691-700.
- German Society for Pediatric Cardiology e.V., ed. Schmaltz AA: Guidelines for diagnosis and therapy in pediatric cardiology. Elsevier GmbH, Urban & Fischer Verlag 2007.
- Knirsch W, et al: New recommendations for antibiotic endocarditis prophylaxis in children in Switzerland. Paediatrica 2009; 20(4): 28-34.
CARDIOVASC 2016; 15(5): 4-9