Interstitial lung disease is relatively uncommon in family practice. Nevertheless, it should be considered in cases of chronic dyspnea, dry cough, and fine-bubbled rales. Early and correct diagnosis is critical.
Interstitial lung diseases (ILD) subsume a large group of diverse rare diseases of the lung parenchyma with similar clinical, radiological, and pathological presentation [1]. The underlying processes of ILD, prognosis, and also therapies vary considerably from entity to entity. On the one hand, we find diseases with a favorable prognosis, but also diseases with an acute course and high mortality. Idiopathic pulmonary fibrosis (IPF), the most common ILD among idiopathic interstitial pneumonias (IIP), shows a comparable course to aggressive malignancies, with a median survival of approximately three years after diagnosis [2].
The treatment of ILD in individual patients depends largely on the disease entity. For some disease patterns, the focus is on exposure abstinence to (inhalative) noxious agents. Immunosuppressive therapies or, in IPF, antifibrotic therapy are used, depending on the entity. Early and correct diagnosis, with particular recognition of reversible factors, is critical for optimal patient counseling and treatment.
Classification of interstitial lung diseases
ILD can be divided into diseases with a known cause and diseases with an unknown cause (idiopathic interstitial pneumonia, IIP) (Fig. 1) . The list of possible causes of ILD is long. We find among the known causes exposures to inorganic or organic dusts, drugs and X-rays. Complicating ILD can occur in many rheumatologic diseases. ILD may represent the initial manifestation of an underlying rheumatologic disease before such a disease can be detected using standard diagnostic criteria. To address this situation, the term “interstitial pneumonitis with autoimmune features” (IPAF) was introduced in 2015 [3].
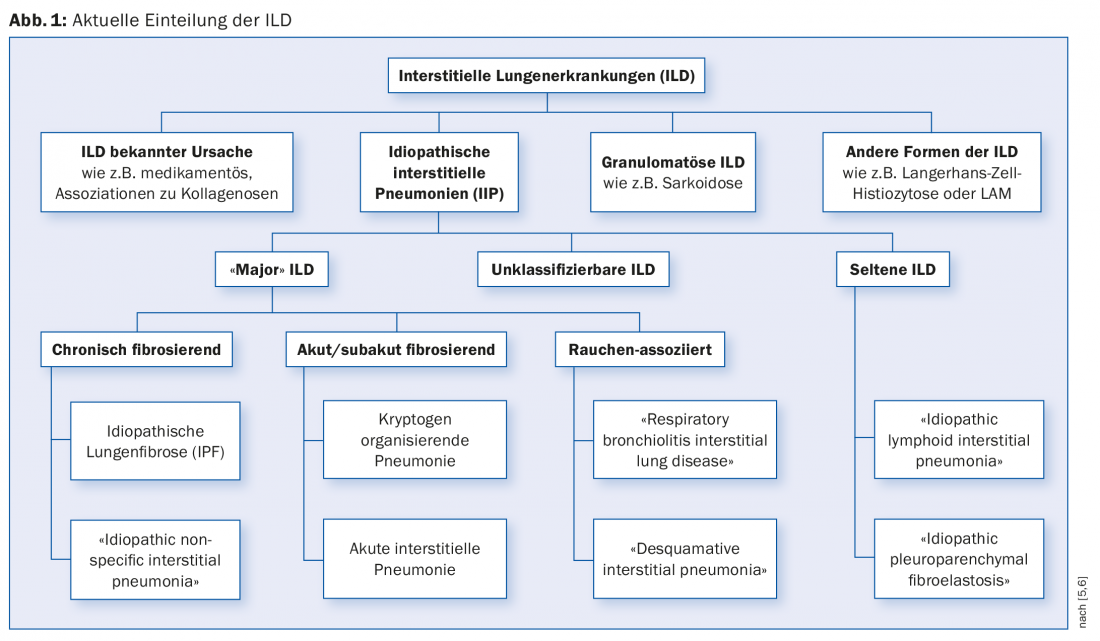
The cause of IIP is – as the name suggests – unclear. “Major” IIP are classified by clinical course into chronic fibrosing, acute/subacute fibrosing, and smoking-associated disease.
The designation “idiopathic” is partly at odds with the pathophysiology in the group of smoking-associated diseases, and a categorization outside the IIP is therefore discussed. Smoking causes histologically detectable changes (“respiratory bronchiolitis”). Susceptible individuals may develop ILD (“respiratory bronchiolitis interstitial lung disease” [RB-ILD] and “desquamative interstitial pneumonia”) [4].
Epidemiology of ILD
ILD are rare diseases, accordingly there are few epidemiological data. A large epidemiologic study in 1994 found an incidence of 31.5 cases per 100,000/year in men, and 26.1 cases per 100,000/year in women [7]. IPF, which is clustered in older men, represents the most common entity among IILDs. Epidemiological data for IPF in Switzerland are not available. Estimates range from 100 to 5000 patients with IPF in Switzerland (prevalence 1.25-63 cases/100,000) [8]. Age at initial diagnosis of ILD varies among the different disease entities. While the incidence of IPF increases markedly with age, sarcoidoses, Langerhans cell histiocytoses, or, in women, lymphangioleiomyomatoses (LAM) are more common in young adulthood.
Diagnosis of ILD
ILD is diagnosed by considering clinical, radiological, pulmonary function, laboratory chemistry and, depending on the situation, cytological/histological findings. The diagnosis and the therapeutic strategy for the individual patient are optimally determined in an interdisciplinary setting consisting of clinicians (pulmonologists), radiologists and pathologists (ILD board). It has been shown that a significant qualitative improvement in diagnostics can be achieved [9].
Clinic: The clinic of ILD is often nonspecific with slowly increasing dyspnea and usually a dry cough. Clinical examination often reveals basal-emphasized, fine-bubble rales and, particularly in advanced disease, decreased oxygen saturation (especially during exercise) and signs of chronic hypoxemia such as watch-glass nails and drumstick fingers. It is important to consider the presence of ILD early in the differential diagnosis of patients with these symptoms.
Often, a careful history provides us with crucial clues as to the entity of ILD. The duration of the symptomatology, but also possible environmental influences including a detailed occupational history are important. The identification of potentially reversible environmental influences is of great importance. Recognition of potential aeroallergens that may trigger hypersensitivity pneumonitis (exogenous allergic alveolitis, EAA) is elementary for both diagnostic purposes and crucial exposure scarcity. Most commonly, we find bird keeping, contact with agriculture, or exposure to molds. Smoking represents a potentially reversible trigger in smoking-associated ILD. Exposure to asbestos, in addition to asbestos-associated pleural disease, can also lead to asbestos-associated pulmonary fibrosis. Symptoms of an underlying rheumatologic disease should be specifically sought. Many medications can cause ILD as a side effect. At this point, we would like to refer to the homepage www.pneumotox.com, which is very helpful in everyday life and provides information on known pulmonary toxic side effects.
Family history should be obtained, in up to 20% of all ILD cases there is a familial form with corresponding gene mutations [10].
Imaging: conventional chest radiography often already suggests interstitial lung disease with increased reticular and/or nodular lung markings.
Computed tomography (CT) of the thorax with “high-resolution” slices plays a central role in the diagnosis of ILD. Based on the nature and arrangement of the changes in relation to the location of the secondary lobule (Fig. 2) and lung topography, different radiological patterns can be distinguished.
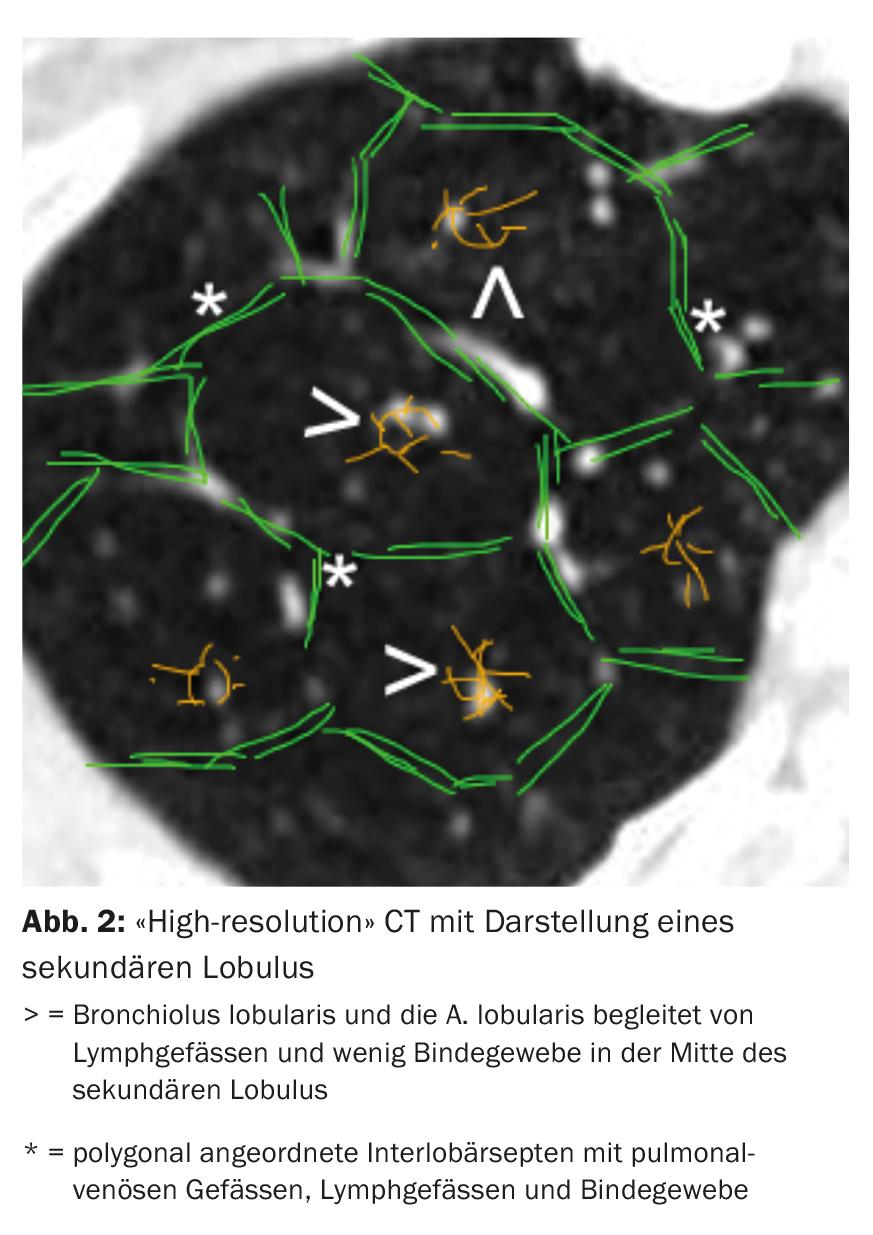
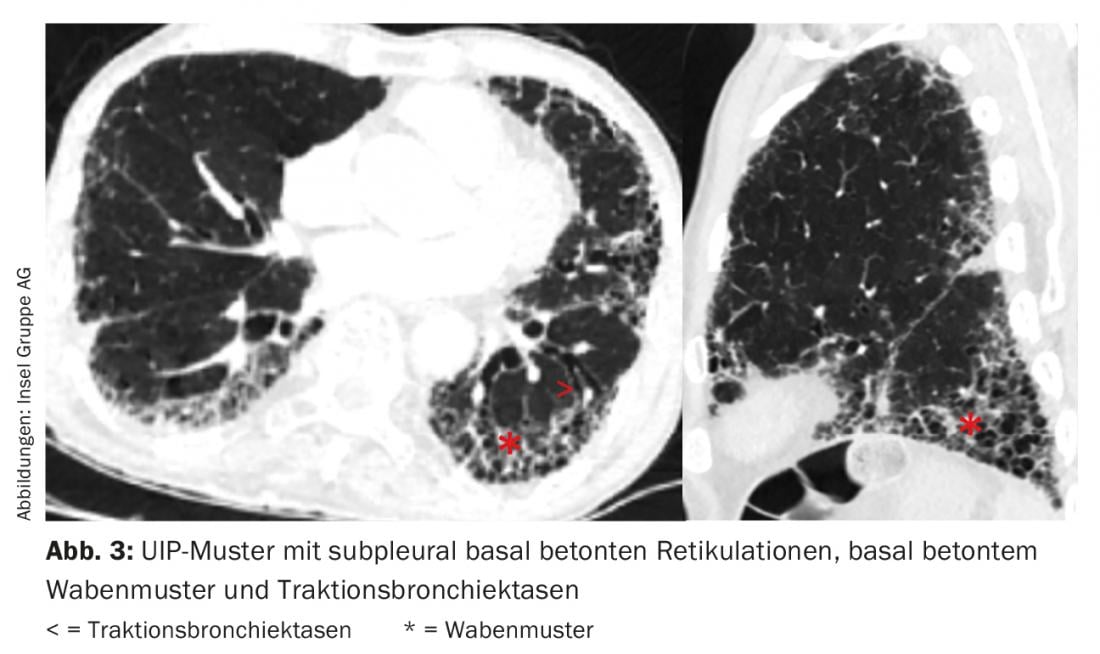
The single radiologic pattern is not specific to an entity of ILD. For example, in IPF, we find a “usual interstitial pneumonia”(UIP) pattern with subpleural basal accentuated reticulations, basal accentuated honeycomb pattern, and traction bronchiectasis (Fig. 3) [11]. However, a UIP pattern may also be found in other entities such as small vessel vasculitis, chronic EAA, ILD in the setting of rheumatoid arthritis, or asbestosis.
Pulmonary function: in ILD we usually find restriction and impairment of gas exchange (diffusion capacity). As the disease progresses, exercise-induced hypoxemia initially develops, followed by resting hypoxemia.
Forced vital capacity (FVC) is used as an important progression parameter. An FVC drop of ≥10% at 24 weeks increases the risk of mortality for the next 12 months by a factor of 4.8 [12]. However, in addition to the easily measured FVC, diffusion capacity, the 6-minute walk test, and multivariable indices also correlate with prognosis [13].
Laboratory: The laboratory may be helpful in classifying ILD. In addition to a blood count (eosinophilia), renal function, liver and inflammatory values, as well as a serological search for an underlying rheumatologic disease can give us indications.
Invasive workup: As a rule, bronchoscopy with at least bronchoalveolar lavage (BAL) is performed in patients with a new diagnosis of ILD. Increasingly hemorrhagic BAL from portion to portion is typical of alveolar hemorrhage (e.g., small-vessel vasculitis). Cell distribution in the BAL shows characteristic changes in some ILD entities (e.g., eosinophilia in eosinophilic pneumonia, lymphocytosis in sarcoidosis and EAA). Depending on the suspected diagnosis based on clinical and radiologic presentation, mediastinal/hilar lymph node biopsies and/or transbronchial lung biopsies are performed. Tissue fragments from transbronchial lung biopsies are often too small for reliable diagnosis. Larger tissue samples can be obtained with thoracoscopy or, since a few years, bronchoscopically with cryobiopsy.
Therapy
The therapy of ILD differs fundamentally from entity to entity (Fig. 4) . For example, in a few entities, the disease can be decisively influenced by exposure abstinence (smoking cessation, abstinence from triggers of exogenous allergic alveolitis, change of medication). Many ILD are treated with immunosuppression such as diseases associated with collagenoses/vasculitides, (cryptogenic) organizing pneumonias, eosinophilic pneumonias, or sarcoidoses. Immunosuppression is usually performed with systemic corticosteroids, often in combination with a steroid-sparing drug (e.g., azathioprine or mycophenolate).
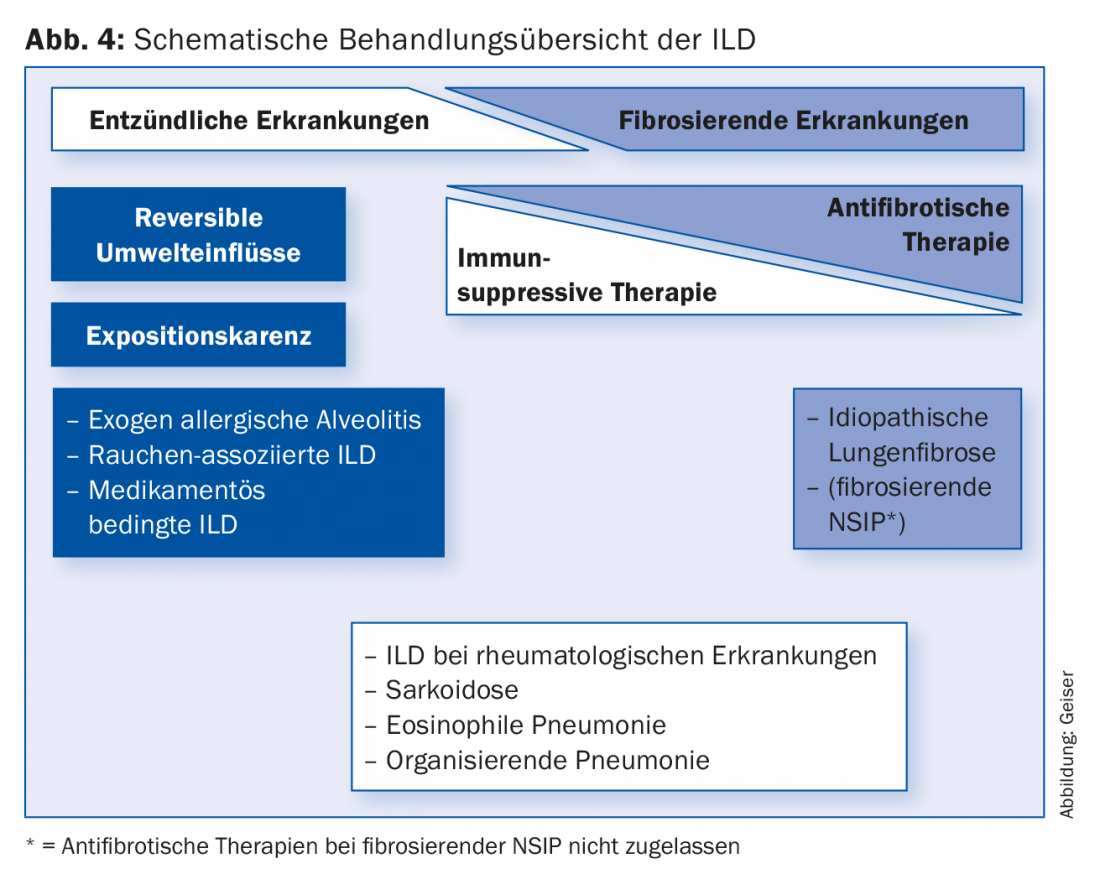
The immunosuppressive therapies commonly used in IPF until a few years ago are no longer recommended [14,15]. After many negative studies with various agents, the two drugs currently approved for the treatment of IPF, pirfenidone (Esbriet®) and nintedanib (Ofev®), have been shown to slow disease progression [16–18]. New data suggest that survival may be favorably affected.
In younger patients with rapidly progressing ILD entities, such as IPF, evaluation for lung transplantation should occur early in the disease course. As ILD progresses, long-term oxygen therapy may be required in hypoxemic patients and further palliative therapies including pulmonary rehabilitation.
Take-Home Messages
- Although interstitial lung disease (ILD) is relatively uncommon in family practice, chronic dyspnea, dry
- cough and fine-bubble rales should be considered as a differential diagnosis.
- Among ILD, we find quite different disease entities with different courses.
- Early and correct diagnosis is critical for optimal patient counseling and treatment.
- Therapy for ILD differs markedly depending on the disease entity and ranges from exposure abstinence to the causative agents to immunosuppression and the use of antifibrotic therapies in idiopathic pulmonary fibrosis (IPF).
- Younger patients with rapidly progressing ILD entity should be evaluated early with regard to lung transplantation.
- In the course, long-term oxygen therapy in hypoxemic patients and further palliative therapies may be required.
Literature:
- Cosgrove G, Schwarz M: Approach to the Evaluation and Diagnosis of Interstitial Lung Disease. In: Schwarz M, King T (eds.): Interstitial Lung Disease. 5th ed. Shelton: People’s Medical Publishing House-USA 2011: 3-34.
- Latsi PI, et al: Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003; 168(5): 531-537.
- Fischer A, et al: An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015; 46(4): 976-987.
- Fraig M, et al: Respiratory bronchiolitis: a clinicopathologic study in current smokers, ex-smokers, and never-smokers. Am J Surg Pathol 2002; 26(5): 647-653.
- American Thoracic Society/European Respiratory Society: International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2002; 165(2): 277-304.
- Travis WD, et al: An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188(6): 733-748.
- Coultas DB, et al: The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med 1994; 150(4): 967-972.
- Funke M, Geiser T: Idiopathic pulmonary fibrosis: the turning point is now! Swiss Med Wkly 2015; 145: w14139.
- Flaherty KR, et al: Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170(8): 904-910.
- Kropski JA, Blackwell TS, Loyd JE: The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J 2015; 45(6): 1717-1727.
- Raghu G, et al: An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183(6): 788-824.
- du Bois RM, et al: Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med 2011; 184(12): 1382-1389.
- Sharp C, Adamali HI, Millar AB: A comparison of published multidimensional indices to predict outcome in idiopathic pulmonary fibrosis. ERJ Open Res 2017; 3(1). DOI: 10.1183/23120541.00096-2016.
- Funke M, et al: Idiopathic Pulmonary Fibrosis in Switzerland: Diagnosis and Treatment. Respiration 2017; 93(5): 363-378.
- Raghu G, et al: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 2015; 192(2): e3-19.
- Noble PW, et al: Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779): 1760-1769.
- Richeldi L, et al: Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22): 2071-2082.
- King TE Jr, et al: A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22): 2083-2092.
HAUSARZT PRAXIS 2017; 12(12): 13-18