Therapy of choice for Ewing’s sarcoma includes neoadjuvant chemotherapy, tumor resection, and postoperative chemotherapy with/without radiotherapy. In this context, interdisciplinary management at an appropriate center is of crucial importance.
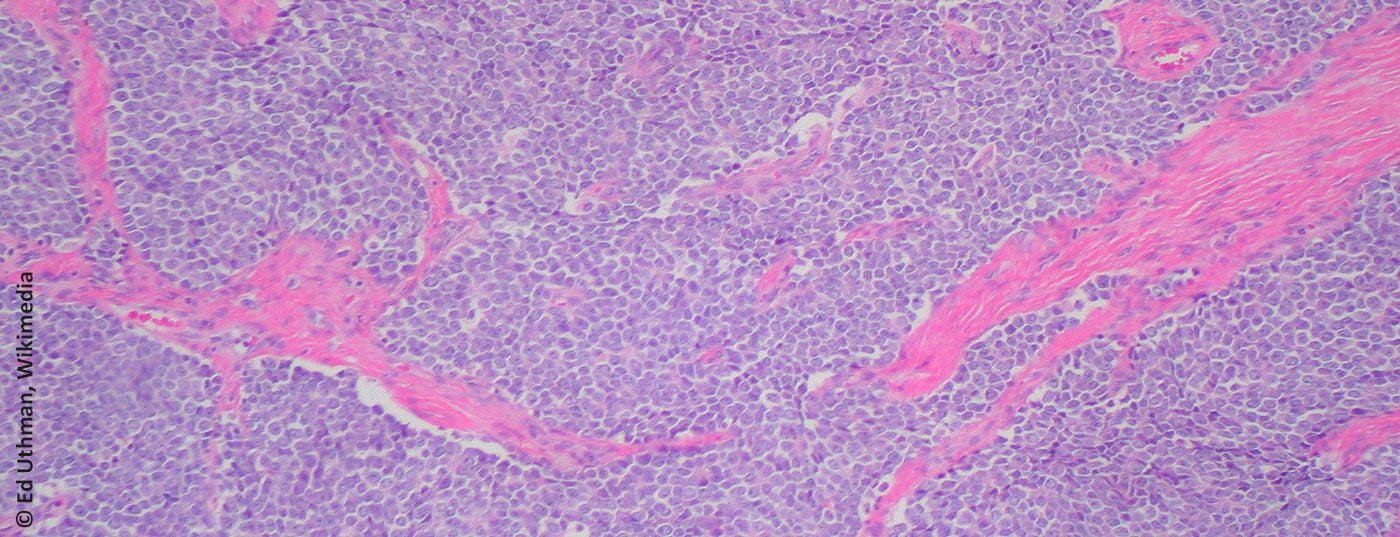
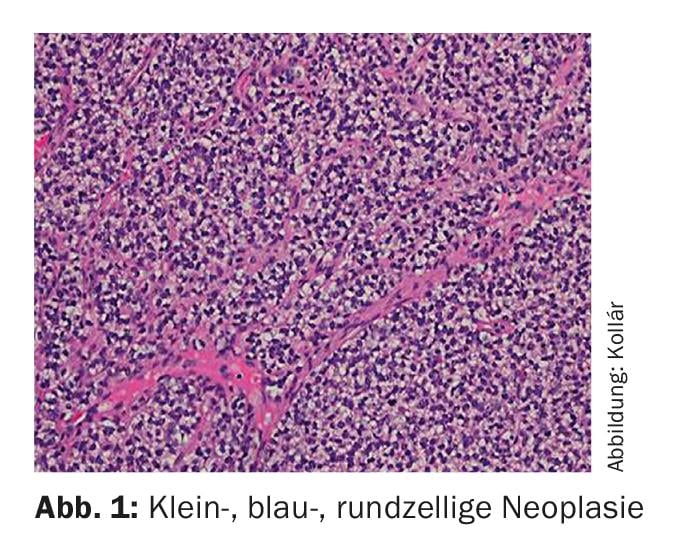
Ewing’s sarcoma was named after James Ewing (American pathologist, 1866-1943). Microscopically, it belongs to the group of small, blue, round cell tumors (Fig. 1) . The cell of origin is not clearly understood, although the presence of neuronal markers suggests a connection with the embryonic neuroectoderm [1]. Diagnosis is usually achieved by molecular diagnostic detection of translocations involving the EWS gene on chromosome 22. t(11;22)(q24;q12) represents the most common translocation (85-95%) [2]. By definition, all Ewing sarcomas are classified as highly malignant (G3) [3].
Epidemiology
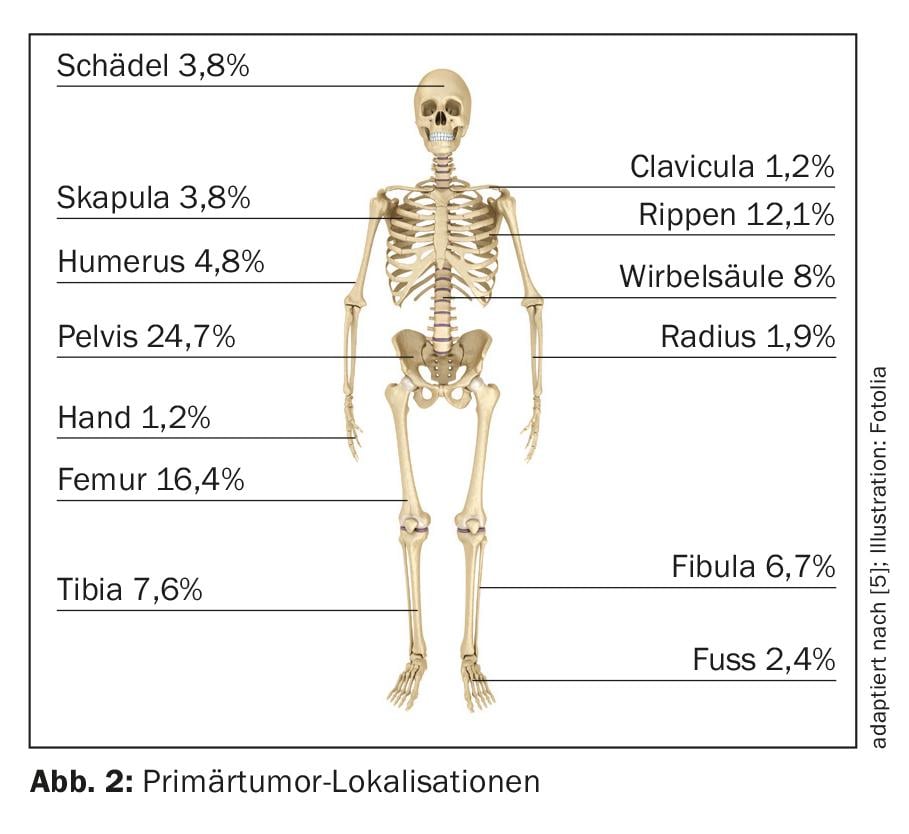
After osteosarcoma, Ewing’s sarcoma represents the second most common primary malignant bone tumor in childhood and adolescence. The median age of onset is 10-15 years, and the annual incidence rate is approximately 3/1,000,000 population [4]. The male sex is slightly more frequently affected (1.5:1). The pelvis (25%) and the diaphyses of the long bones, especially in the femur (approximately 16%) (Fig. 2), are among the most common primary tumor localizations [5]. In 15% of cases, an extraosseous localization can be primarily documented [6].
Clinic
The initial symptomatology is nonspecific. A localized pain and/or a swelling, possibly with consecutive mobility restriction, are in the foreground of the symptoms. In approximately 10-15% of cases, a pathologic fracture is present at initial diagnosis [7], and in 80%, a formally localized tumor stage is present. Due to a very high metastasis rate (>80%) after exclusively local therapy of the primary tumor, pre-existing subclinical metastases must be assumed in almost all cases [8]. Metastasis most commonly occurs pulmonary, osseous, and bone marrow [9].
Diagnostics
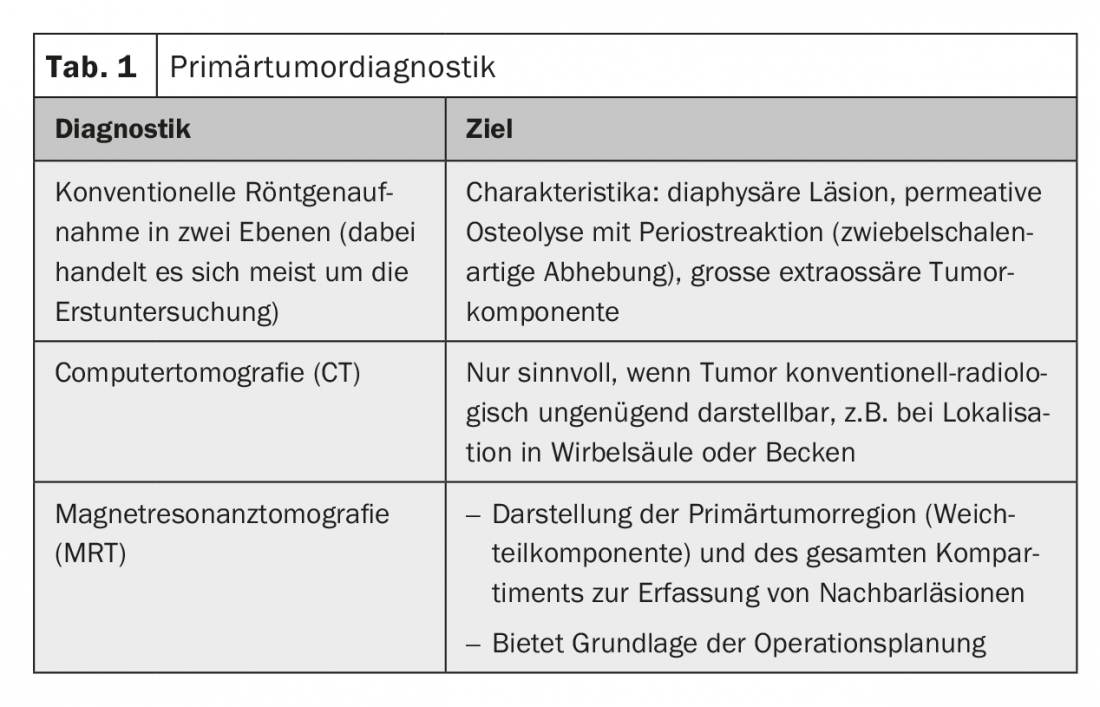
Radiological diagnosis of the primary tumor: Radiological diagnosis of the primary tumor should definitely be performed prebioptically. It is the basis for an assessment of tumor significance and resectability as well as for planning the biopsy (Table 1).
Biopsy: If a malignant soft tissue or bone tumor is suspected, biopsy is mandatory to confirm the diagnosis. The biopsy should be performed with the involvement of a surgeon experienced in the treatment of sarcomas, ideally the future surgeon. The surgical access route must be taken into account here. The gold standard is core needle biopsy.
Staging: After biopsy confirms the diagnosis, the following staging tests should be performed:
- CT thorax (exclusion of pulmonary metastases)
- Skeletal scintigraphy (exclusion of osseous metastases)
- Bone marrow biopsy and aspiration (only indicated if PET/CT is not performed due to low incidence).
- Other imaging modalities depending on clinical complaints.
The value of PET/CT examination in initial staging and as follow-up imaging is currently being investigated in clinical trials. The sensitivity of detecting pulmonary metastasis is lower compared with CT thorax, and the sensitivity of detecting osseous lesions is higher [10,11].
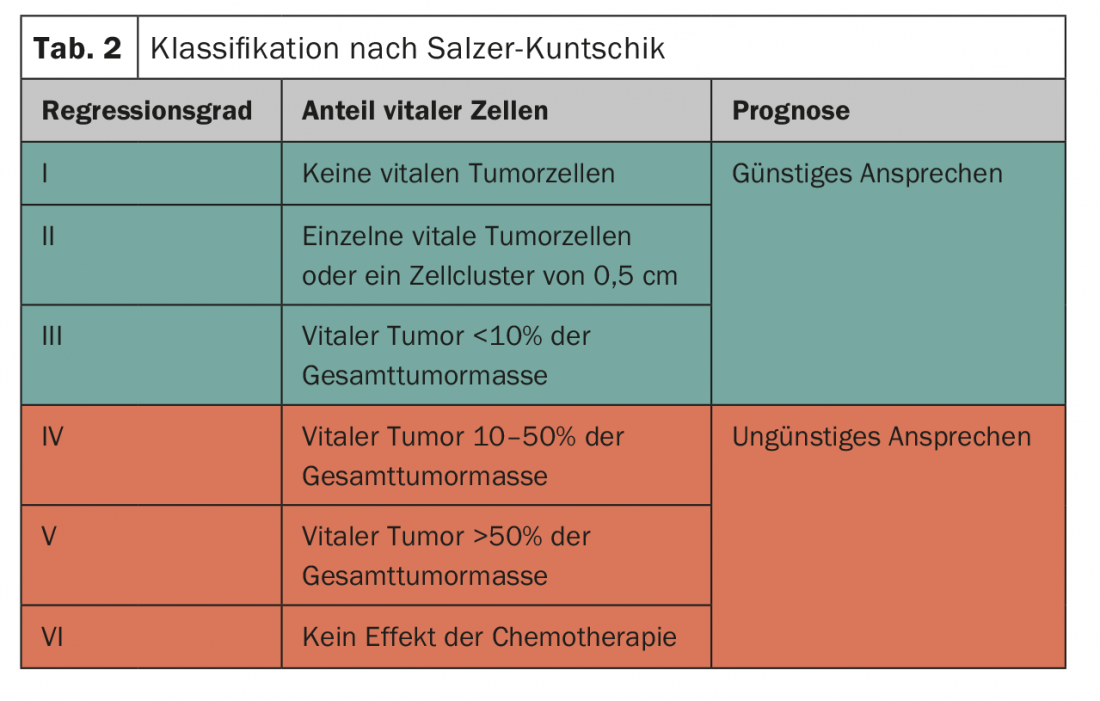
Histological tumor response to system therapy: To assess chemotherapy response, the proportion of vital malignant cells is determined in the definitive resection specimen. The pathologist then classifies the response in German-speaking countries based on this assessment (Tab. 2) [12]. The classification has prognostic significance; it is highly relevant, particularly in the context of the Euro-Ewing studies, for determining the postoperative therapeutic strategy.
Localized stage of disease
Therapy of Ewing’s sarcoma is multimodal and should be discussed in an interdisciplinary manner in a sarcoma center. This always includes local therapy (resection and/or radiotherapy) and chemotherapy. Early prospective randomized trials demonstrated significantly better overall survival with the addition of chemotherapy (10-20% vs. nearly 70% in 5-year event-free survival) [8,13], which is why the current standard of care includes neoadjuvant chemotherapy followed by local therapy of the primary tumor and adjuvant chemotherapy [14]. Patients should be treated under a study protocol when possible.
Surgery: Resection of the primary tumor is the local therapy of choice. The goal is always to achieve complete tumor resection. However, resectability of the primary tumor is sometimes not given depending on the anatomical localization. These primarily include Ewing sarcomas of the spine and pelvis. The modality of therapy must be individualized in these cases. Here, resection with postoperative radiotherapy or radiotherapy alone is targeted [15,16].
Radiotherapy: as radiosensitive tumors, Ewing’s sarcomas show comparable local control rates with radiotherapy as surgically treated patients in certain studies [17]. Definitive radiotherapy is preferred for nonresectable primary tumors according to an incomplete resection followed by postoperative radiotherapy [17]. In cases of marginal/intralesional excision, there is an indication to perform postoperative radiotherapy. The value of additive radiotherapy in cases of insufficient histologic response to chemotherapy (but complete tumor resection) is unclear. In principle, radiotherapy can also be given preoperatively [18].
System therapy: The international standard is combination chemotherapy. The most effective chemotherapeutic agents include alkylating agents (ifosfamide, cyclophosphamide), anthracyclines (doxorubicin), and etoposide, vincristine, and actinomycin. The VIDE regimen (vincristine, ifosfamide, doxorubicin, etoposide) analogous to the Euro-Ewing protocol in 1999 and 2008 in Europe and the VDC/IE regimen (VIDE agents + cyclophosphamide) in America often serve as templates for induction chemotherapy. The addition of ifosfamide and etoposide to VDC was associated with a significant prolongation of 5-year event-free survival in the randomized IESS-III trial (69% vs. 54%) [19]. The direct comparison between the schemes is tested in the most recent Euro-Ewing study 2012.
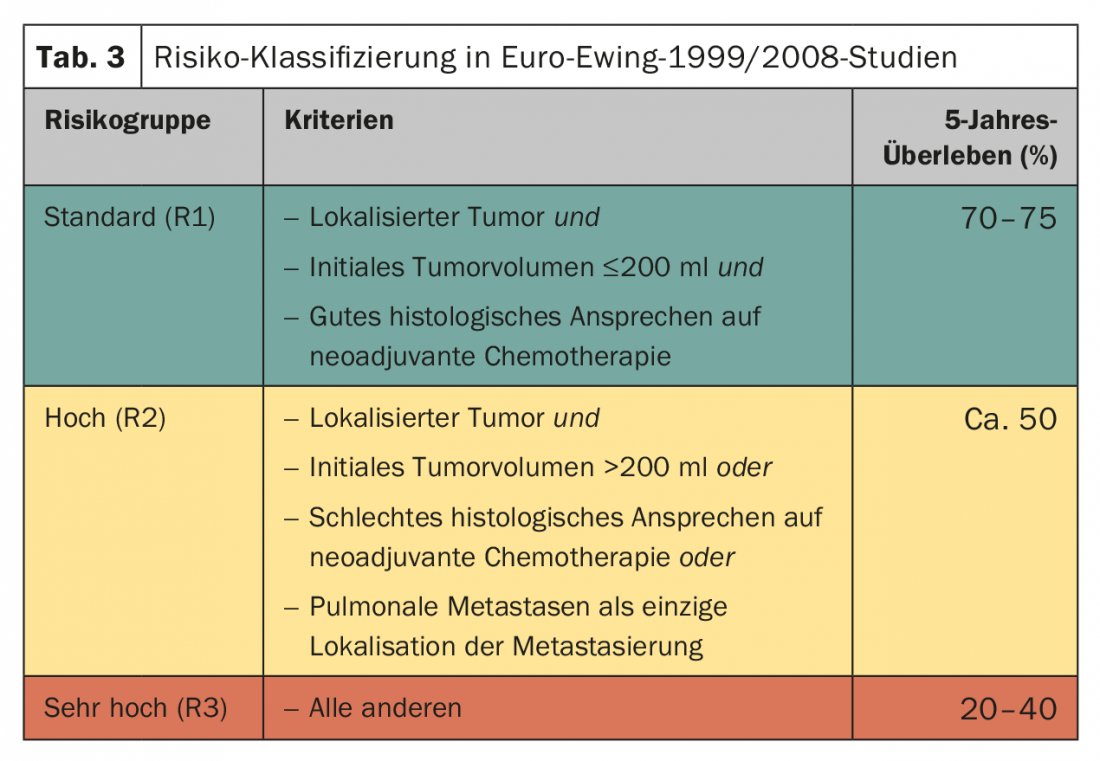
Local therapy usually follows six cycles of neoadjuvant chemotherapy. The postoperative therapy is planned depending on the risk constellation (Tab. 3) . The standard is eight cycles of chemotherapy according to VAI or VAC. The Euro-Ewing 1999 trial randomly compared the efficacy of VAC (cyclophosphamide) and VAI (ifosfamide) in the R1 risk group. Basically, the equivalence of the schemes was confirmed, with the male gender seeming to benefit more from VAI (HR 1.34; 95% CI 0.96-1.86) [20]. The total duration of therapy is approximately ten to twelve months. In the R2 risk group (localized tumor stage, poor histologic tumor response, tumor volume >200 ml), the value of melphalan-containing high-dose therapy with autologous stem cell transplantation was randomized. Here, a significant improvement in overall survival was achieved by high-dose chemotherapy with busulfan/melphalan (77.8 vs. 69.9%, HR 0.60 [0,39–0,92], p=0.019) [21].
Metastatic disease stage
In the metastatic stage, Ewing’s sarcoma is generally responsive to the same chemotherapeutic agents used in the localized stage. Depending on the number and especially localization, there is curative potential despite the presence of hematogenous metastases. For isolated pulmonary/pleural metastasis, the cure rate is up to 40%, for bone and bone marrow metastasis, the cure rate is approximately 20-25%, and for a combination of these sites, the cure rate is 15% [22]. Such results are achieved in particular with consistent local therapy of all metastases.
Pulmonary/pleural metastases: If persistent pulmonary metastases are resected after induction chemotherapy, this seems to be associated with an improved prognosis [23] – accordingly the recommendation to surgically remove radiologically visible pulmonary metastases during the course. Because more foci are often seen intraoperatively than preoperative staging had objectified, resection should be performed by open surgery [24].
Likewise, according to retrospective analyses, lung parenchymal irradiation (between 15 and 20 Gy) is associated with a more favorable prognosis in isolated pneumo-pleural metastasis [25,26]. Thus, the indication should be considered in complete remission after chemotherapy and after resection of all pulmonary foci.
Bone and bone marrow metastases: In osseous and/or bone marrow metastasis, prognosis is poor despite the potentially curative goal of therapy. In case of oligometastatic involvement, it is recommended to consider local therapy options. Radiotherapy is the main method used here.
High-dose chemotherapy with autologous stem cell transplantation: The value of high-dose chemotherapy with autologous stem cell support has long been controversial in the literature. A prospective non-randomized study showed exceptional results with 5-year event-free survival of up to 52%. However, other publications could not support these results [27,28]. Recently, the results of R2 stratification were reported in the context of Euro-Ewing 1999. In patients with pulmonary metastases, high-dose chemotherapy without lung irradiation failed to demonstrate benefit compared with conventional chemotherapy with lung irradiation [29]. The results of the R3 risk group are still pending.
Treatment of recurrences
The most common recurrences are found in the first five years after initial diagnosis, but late recurrences are also not uncommon [30]. The prognosis of recurrences within the first two years is very poor, whereas later recurrences show long-term survival in approximately 15-20% [31]. Depending on the time of recurrence, localization and number of tumor manifestations, and prior therapy, the therapy of choice results. Local recurrences and isolated pulmonary metastases are usually treated locally, i.e., with resection and/or radiotherapy [32]. In the case of extensive recurrence, the initiation of systemic therapy is again indicated, although there is no standard regimen for this. If the response to the initial therapy is good and, above all, prolonged, a repetition of this can be evaluated. The cumulative doxorubicin dose must not be disregarded. In general, there is a tendency to intensify chemotherapy using high-dose therapy, although the evidence in this regard is limited [33]. Chemotherapy regimens used include topotecan/cyclophosphamide, irinotecan/temozolomide, gemcitabine/docetaxel, high-dose infosfamide, and platinum-containing chemotherapy with etoposide [34–36]. Molecularly targeted therapies (e.g. IGF-1 and PARP inhibitors) as well as immunotherapeutic approaches are currently being investigated in trials.
Aftercare
Follow-up is aimed at early detection of recurrences and monitoring of late toxicities. Currently, there is a lack of prospective data showing a survival benefit from regular follow-up examinations. Attempts are made to address the increased likelihood of recurrence in the first two to three years with the follow-up intervals. Recommendations can be found, for example, in the National Comprehensive Cancer Network (NCCN) guidelines.
The most common are secondary neoplasms (cumulative incidence 9%), endocrinopathies incl. Infertility, cardio-, nephro-, and neurotoxicity, pulmonary toxicity, and locoregional functional impairment in the context of the local therapy that took place as therapeutic late toxicities on [37]. Details and recommendations on late effects monitoring can be found at www.survivorshipguidelines.org.
Take-Home Messages
- Diagnosis and therapy of Ewing’s sarcoma are performed in an interdisciplinary manner at a sarcoma center. Radiological diagnosis of the primary tumor should be performed prebiopsy. therapy of choice is a neoadjuvant
- Chemotherapy, a tumor resection, and postoperative chemotherapy with/without radiotherapy.
- The tumor should be resected by an experienced surgeon in sarcomoncology.
- Even in metastatic tumor stage, the therapeutic goal is curative.
Literature:
- Lipinski M, et al: Neuroectoderm-associated antigens on Ewing’s sarcoma cell lines. Cancer research 1987; 47: 183-187.
- de Alava E, Gerald WL: Molecular biology of the Ewing’s sarcoma/primitive neuroectodermal tumor family. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2000; 18: 204-213.
- Fletcher CDM BJ, Hogendoorn P, Mertens F (eds.): WHO Classification of Tumours of Soft Tissue and Bone (IARC WHO Classification of Tumours). 4th ed. 2013.
- Esiashvili N, Goodman M, Marcus RB Jr: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. Journal of pediatric hematology/oncology 2008; 30: 425-430.
- Cotterill SJ, et al: Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2000; 18: 3108-3114.
- Applebaum MA, et al: Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 2011; 117: 3027-3032.
- Widhe B, Widhe T: Initial symptoms and clinical features in osteosarcoma and Ewing sarcoma. The Journal of bone and joint surgery American volume 2000; 82: 667-674.
- Nesbit ME Jr, et al: Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 1990; 8: 1664-1674.
- Applebaum MA, et al: Clinical features and outcomes in patients with Ewing sarcoma and regional lymph node involvement. Pediatric blood & cancer 2012; 59: 617-620.
- Franzius C, et al: FDG-PET for detection of pulmonary metastases from malignant primary bone tumors: comparison with spiral CT. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO 2001; 12: 479-486.
- Franzius C, et al: FDG-PET for detection of osseous metastases from malignant primary bone tumours: comparison with bone scintigraphy. European journal of nuclear medicine 2000; 27: 1305-1311.
- Salzer-Kuntschik M, et al: Morphological grades of regression in osteosarcoma after polychemotherapy – study COSS 80. Journal of cancer research and clinical oncology 1983; 106 Suppl: 21-24.
- Burgert EO Jr, et al: Multimodal therapy for the management of nonpelvic, localized Ewing’s sarcoma of bone: intergroup study IESS-II. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 1990; 8: 1514-1524.
- Womer RB, et al: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children’s Oncology Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2012; 30: 4148-4154.
- Vogin G, et al: Local control and sequelae in localised Ewing tumours of the spine: a French retrospective study. Eur J Cancer 2013; 49: 1314-1323.
- Puri A, et al: Results of surgical resection in pelvic Ewing’s sarcoma. Journal of surgical oncology 2012; 106: 417-422.
- La TH, et al: Radiation therapy for Ewing’s sarcoma: results from Memorial Sloan-Kettering in the modern era. International journal of radiation oncology, biology, physics 2006; 64: 544-550.
- Krasin MJ, et al: Definitive irradiation in multidisciplinary management of localized Ewing sarcoma family of tumors in pediatric patients: outcome and prognostic factors. International journal of radiation oncology, biology, physics 2004; 60: 830-838.
- Grier HE, et al: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. The New England journal of medicine 2003; 348: 694-701.
- Le Deley MC, et al: Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2014; 32: 2440-2448.
- Whelan J, et al: Efficacy of busulfan-melphalan high dose chemotherapy consolidation (BuMel) in localized high-risk Ewing sarcoma (ES): results of EURO-EWING 99-R2 randomized trial (EE99R2Loc). Journal of Clinical Oncology 2016; 34(15) Suppl: 11000-11000.
- Paulussen M, et al: Ewing’s tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing’s Sarcoma Studies patients. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 1998; 16: 3044-3052.
- Letourneau PA, et al: Resection of pulmonary metastases in pediatric patients with Ewing sarcoma improves survival. Journal of pediatric surgery 2011; 46: 332-335.
- Ladenstein R, et al: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2010; 28: 3284-3291.
- Dunst J, Paulussen M, Jurgens H: Lung irradiation for Ewing’s sarcoma with pulmonary metastases at diagnosis: results of the CESS-studies. Radiotherapy and Oncology: Organ of the German Rontgen Society [et al] 1993; 169: 621-623.
- Paulussen M, et al: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO 1998; 9: 275-281.
- Oberlin O, et al: Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Societe Francaise des Cancers de l’Enfant. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2006; 24: 3997-4002.
- Ladenstein R, et al: Impact of megatherapy in children with high-risk Ewing’s tumours in complete remission: a report from the EBMT Solid Tumour Registry. Bone marrow transplantation 1995; 15: 697-705.
- Dirksen U, et al: Efficacy of busulfan-melphalan high dose chemotherapy consolidation (BuMel) compared to conventional chemotherapy combined with lung irradiation in ewing sarcoma (ES) with primary lung metastases: results of EURO-EWING 99-R2pulm randomized trial (EE99R2pul). Journal of Clinical Oncology 2016; 34(15) Suppl: 11001-11001.
- Weston CL, et al: Establishing long-term survival and cure in young patients with Ewing’s sarcoma. British journal of cancer 2004; 91: 225-232.
- Stahl M, et al: Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatric blood & cancer 2011; 57: 549-553.
- Bacci G, et al: Metachronous pulmonary metastases resection in patients with Ewing’s sarcoma initially treated with adjuvant or neoadjuvant chemotherapy. Eur J Cancer 1995; 31A: 999-1001.
- Rasper M, et al: The value of high-dose chemotherapy in patients with first relapsed Ewing sarcoma. Pediatric blood & cancer 2014; 61: 1382-1386.
- Hunold A, et al: Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatric blood & cancer 2006; 47: 795-800.
- van Maldegem AM, et al: Etoposide and carbo-or cisplatin combination therapy in refractory or relapsed Ewing sarcoma: a large retrospective study. Pediatric blood & cancer 2015; 62: 40-44.
- Casey DA, et al: Irinotecan and temozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatric blood & cancer 2009; 53: 1029-1034.
- Ginsberg JP, et al: Long-term survivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. Journal of the National Cancer Institute 2010; 102: 1272-1283.
InFo ONCOLOGY & HEMATOLOGY 2018; 6(5): 13-16.