Hematopoietic stem cell myeloproliferative neoplasia is predominantly caused by somatic mutations of the JAK2 gene and resulting clonal autonomous hematopoiesis. The risk of myelofibrosis or leukemic transformation are prognostic.
Polycythemia vera (PV) is currently classified by the World Health Organization (WHO) in the major category of myeloproliferative neoplasms (MPNs) [1,2]. According to current knowledge, PV is a disease of hematopoietic stem or progenitor cells predominantly caused by somatic mutations in the JAK2 gene with resulting clonal myeloproliferation [3]. Rare familial clusters have been described, with these having germline mutations in the erythropoietin receptor (EPOR) [4]. The incidence of PV in Europe is 0.4-2.8% population per year. The mean age of onset is 60-65 years, and women and men are affected in approximately equal numbers [5,6]. Median survival is 14 years in elderly PV patients (>60 years) and 24 years in those younger than 60 years [7].
Clinic
In the blood picture, the disease manifests itself by an increased production of erythrocytes (erythrocytosis) independent of physiological regulatory mechanisms. Leukocytosis and thrombocytosis due to increased megakaryopoise often accompany the disease. In the course of the disease, progressive fibrotic changes in the sense of myelofibrosis lead to a so-called “spent phase” with reduced hematopoiesis in up to 10% of all PV patients [8]. Leukemic transformation is diagnosed in up to 5% of PV cases after 20 years of disease progression [7,9]. Prognostic risk factors for myelofibrosis, leukemic transformation, and reduced overall survival include advanced patient age, leukocytosis, thrombocytosis, and abnormal karyotype [10].
Furthermore, of crucial prognostic relevance is the occurrence of venous and arterial thromboembolic events in 20-50% of all PV patients. Pathophysiologically, these underlie increased blood viscosity and resulting rheologic changes, as well as inflammatory, procoagulant, and microvascular stimuli [11,12]. Two risk groups for recurrent thrombosis are distinguished: High-risk in patients >60 years and positive history for thrombotic events and a low-risk group in the absence of both factors [13]. Hypertension is an independent risk factor for the occurrence of arterial thrombi.
Clinically, PV is manifested mainly by fatigue (up to 84.9%) [14] and pruritus (in about 40%) [15]. Other clinical symptoms include reddened facial skin, blue-red skin and mucous membranes, head pressure, headache, and hypertension. Microcirculatory disturbances often result in characteristic clinical symptoms (e.g., visual disturbances, paresthesias, erythromelalgia, pectanginal symptoms) [16]. Abdominal venous thrombosis and sinus vein thrombosis are other complications with serious and often life-threatening consequences [17,18]. Paradoxically, in rare cases with extreme thrombocytosis (>1000×109/L), there is also an increased tendency to bleed. The cause is a reduction in Von Willebrand factor (VWF) in the sense of an acquired consumption syndrome. Pathobiologically, both increased ADAMTS13 activity and increased platelet sensitivity and resulting inadequate platelet activation play a major role [19,20]. Splenomegaly is found in approximately one-third of all affected individuals and may be associated with pain and infarction.
Pathobiology of PV
PV is characterized by clonal myeloproliferation due to pathological changes in hematologic stem cells [3]. In almost all patients with PV, a mutation of Janus kinase 2 (JAK2) is found, which is located at gene locus 9p24, with 96% of all cases having an activating somatic mutation in exon 14 (JAK2V617F) [21] and less frequently (approximately 3%) in exon 12 [22]. To date, no significant differences in clinical course depending on the particular mutation have been demonstrated [23].
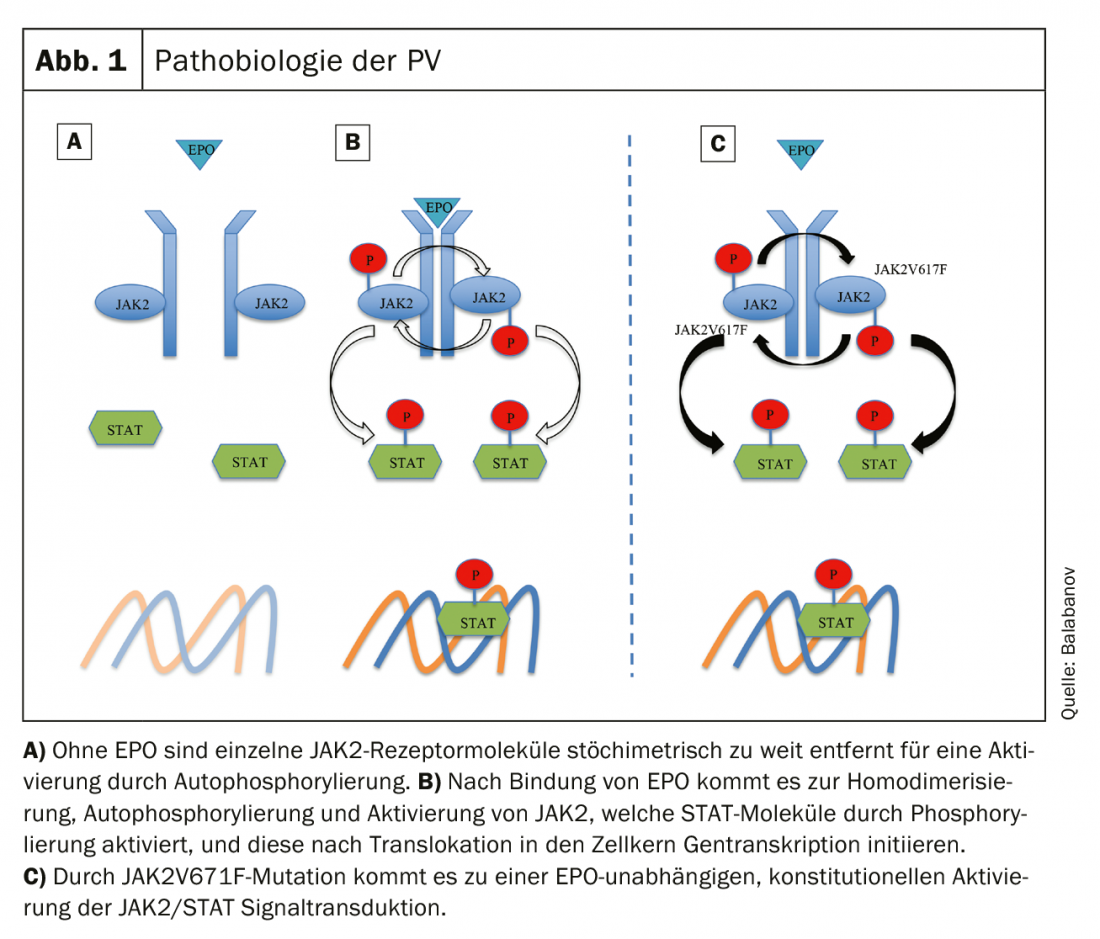
Under physiological conditions, binding of erythropoietin (EPO) to the EPO receptor (EPOR) leads to a conformational change (receptor homodimerization) with resulting autophosphorylation and activation of JAK. Activated JAK molecules phosphorylate and subsequently activate STAT (signal transducer and activator of transcription) molecules, which act as transcription factors to trigger hematopoietic growth signals after translocation to the nucleus [24]. Activating JAK2V617F mutation results in EPO-independent constitutional JAK/STAT activation and uncontrolled cell proliferation (Fig. 1) [25]. Other genetic alterations such as dysregulation of the apotosinhibitor Bcl-x or the transcription factor NF-E2 have been described but appear to be secondary consequences of a JAK2 mutation [26].
JAK2V617F mutations are more commonly found in elderly patients and are associated with panmyelocytosis, myelofibrosis, and clinical symptoms of pruritus [22,23]. No association with overall survival or transformation risk has been demonstrated [3]. The frequency of mutant alleles also does not appear to affect survival or the frequency of leukemic transformation. In a Mayo Clinic study, targeted deep sequencing demonstrated that more than half (53%) of the PV patients studied had additional mutations besides JAK2 [28]. Mutations in TET2 (22%), ASXL1 (12%) SH2B3 (9%) were detected most frequently (other mutations showed up in SRSF2, IDH2, TP53). In particular, mutations in ASXL1, SRSF2, and IHD2 showed a prognostic effect independent of other predictors (median survival 7.7 years with vs. 16.9 years without mutations). An abnormal karyotype is found in 15% of patients at initial diagnosis, with the most common cytogentic alterations detected being -Y, +8, +9, del(20q) and 1q+ [27].
Diagnostics
A detailed medical history and physical examination form the basis of the diagnosis. The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF) questionnaire allows a structured and complete assessment of relevant symptoms [29]. In particular, due to prognostic and treatment relevance, attention should be paid to history of thrombotic events, thrombosis stigmata, hypertension, and patient age. Abdominal ultrasonography is recommended to evaluate for splenomegaly.
In addition to a differential blood count and coagulation status, the EPO concentration must be determined in the laboratory. The final diagnosis can only be made on the basis of a bone marrow biopsy.
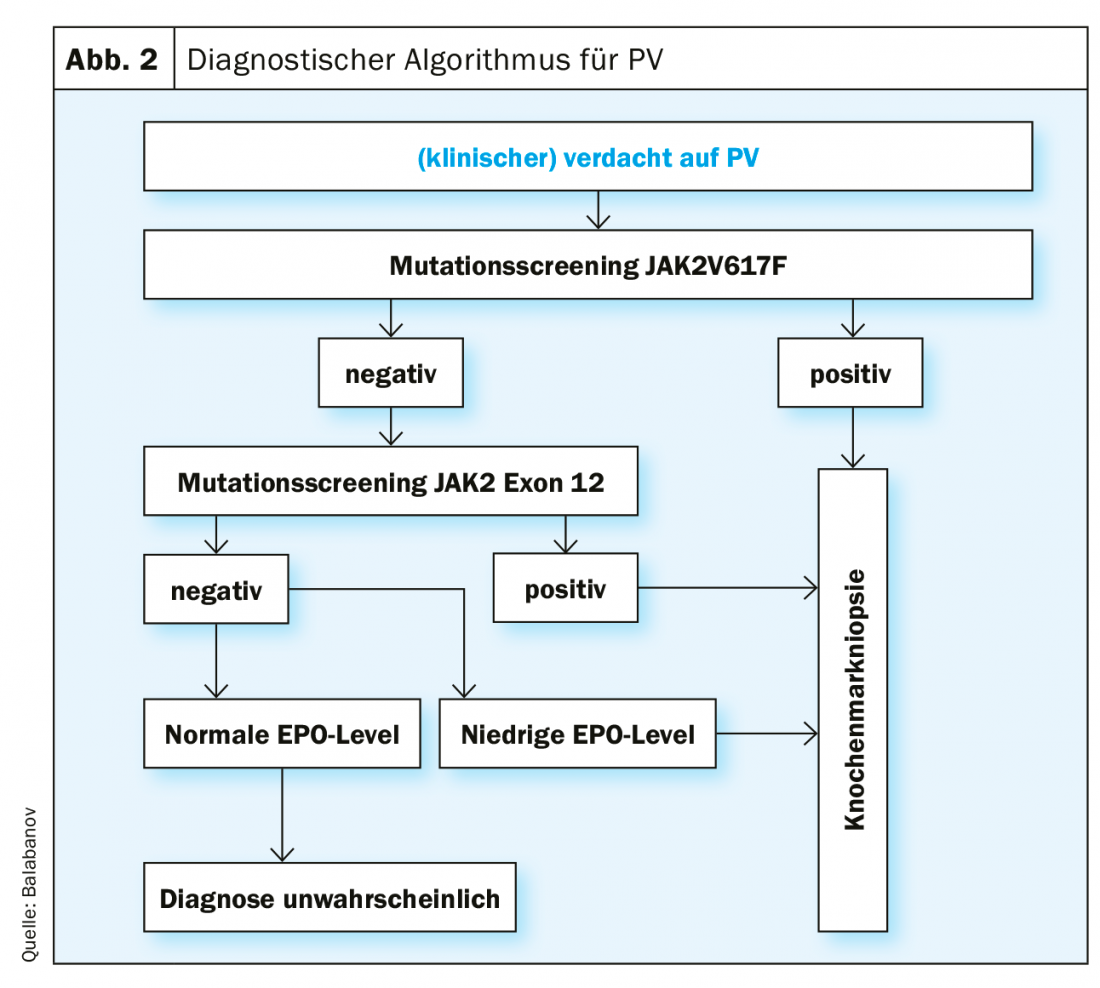
The diagnostic criteria according to the current (2016) WHO classification are shown in Table 1. To make a diagnosis of PV, all major criteria or two major criteria and the minor criterion must be met. The detection of a JAK2V617F mutation is extremely reliable with a test sensitivity of 97% and a specificity of nearly 100%. In case of negative JAK2V617F mutation status but decreased serum EPO level, additional mutation analysis in exon 12 of the JAK2 gene is indicated. Because of the prognostic relevance for both overall survival and leukemia-free survival, karyotype determination should be performed when appropriate. Furthermore, mutational analysis by next generation sequencing (NGS) is useful because mutations in ASXL1, SRSF2, and IDH2 are of prognostic relevance. However, NGS is not currently a standard investigation for every patient with PV at the time of diagnosis. However, a recently published landmark study demonstrated that the comprehensive integration of genomic and clinical variables enables personalized risk stratification with therapeutic implications [30] (https://cancer.sanger.ac.uk/mpn-multistage).

An algorithm for the diagnostic procedure in suspected PV, based on the current WHO diagnostic criteria, is shown in Figure 2. Special functional tests of VWF are indicated to clarify an increased bleeding tendency (e.g., ristocitin cofactor activity).
Therapy
With a 10-year survival rate of more than 75%, as well as a relatively low risk of myelofibrosis (<10%) and malignant transformation in terms of leukemic transformation (<5%) [8], the main goal of treatment of PV is to prevent thrombo-hemorrhagic complications and to alleviate the symptoms described above.
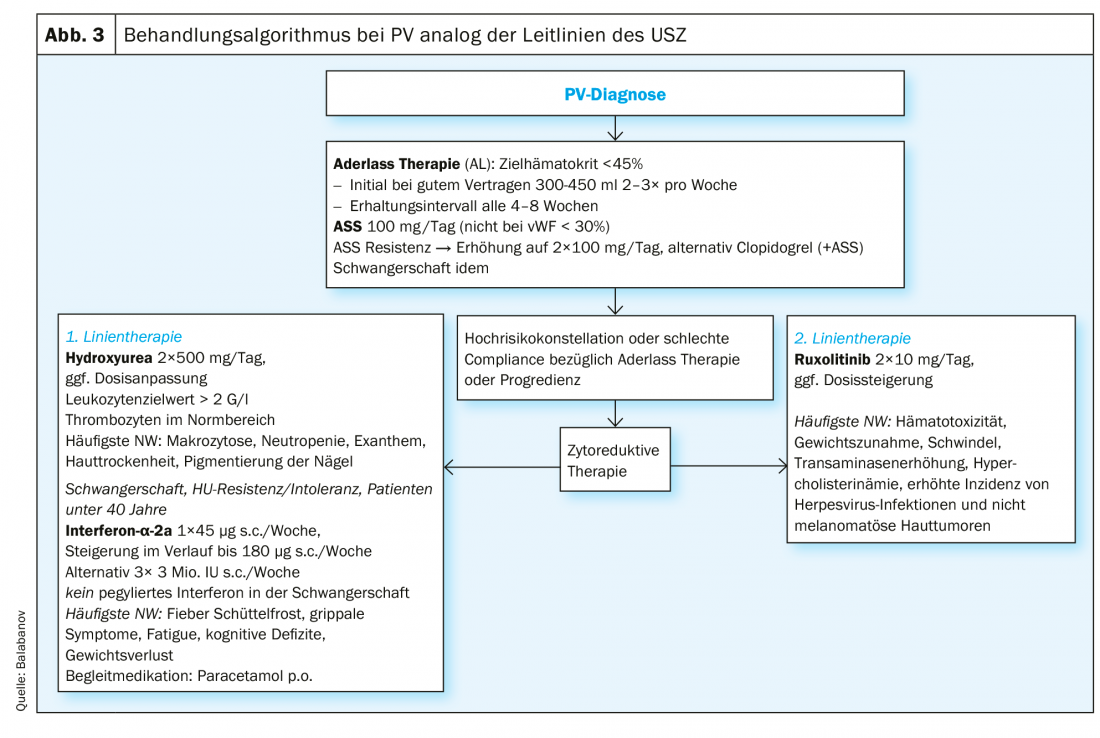
Low-risk patients (age <60 years, negative history for thrombotic events): We recommend consistent phlebotomy therapy, aiming for a target hematocrit <45%, as this target value is superior to higher values (45-50%) in terms of event-free survival [31]. A positive effect on disease symptoms, on the other hand, has not yet been documented [34]. Furthermore, anti-aggregative therapy with aspirin (ASA) is recommended to prevent venous and arterial thrombi [31–33]. Treatment with ASA appears to have a beneficial effect on associated symptomatology by reducing microvascular dysfunction [35]. In the case of aspirin-resistant symptoms, twice-daily administration or alternatively the use of clopidogrel alone or in combination with ASA may be considered [36, 37]. If necessary, a platelet function test (platelet aggregometry, PFA-100) is indicated to distinguish congenital defects from iatrogenic causes [38]. Caution in the use of aspirin is advised in view of the possible increased bleeding tendency in PV patients with concomitant extreme thrombocytosis (>1000×109/L).
High-risk patients (age >60 years, positive history of thrombotic events): In addition to the therapeutic measures mentioned above, we recommend cytoreductive treatment in this patient group, with the primary goal being normalization of the hematocrit. However, a reduction of the frequently accompanying thrombocytosis and leukocytosis should also be aimed for. The current recommendations for substances for first-line therapy according to the European LeukemiaNET (ENL) are hydroxyurea and interferon α (INFα) (pegylated form) [39]. Treatment failure under hydroxyurea (Table 2) in terms of resistance or intolerance occurs in approximately 24% of patients [7]. Busulfan has historically been used with good success in second-line therapy. Today, its use is essentially obsolete due to suspected leukemogenic potential, with no solid data available with regard to the increased incidence of leukemias under busulfan [40].

The JAK2 inhibitor ruxolitinib is approved as second-line therapy for PV and showed positive results in myeloproliferation, need for phlebotomy, and PV-associated symptoms such as fatigue and pruritus while being well tolerated [41–44]. For refractory pruritus, benefit from off-label use of SSRIs [45], INFα [46], and phototherapy [47] has been described. A summary of the treatment algorithm of the University Hospital Zurich (USZ) can be found in Figure 3. Allogeneic bone marrow transplantation or peripheral blood stem cell transplantation should be considered as a curative approach only in exceptional cases in younger patients with a complication-rich course or a disease progression that cannot be controlled by other measures [48]. Because of the increased cardiovascular risk, all patients should be encouraged to reduce cardiovascular risk in terms of primary prevention (exercise, weight control, diet).
Pregnancy: PV is not a contraindication to pregnancy, but these are always considered high-risk pregnancies with an increased rate of spontaneous abortions compared to the normal population [49]. In principle, we recommend care at a specialized center with appropriate hematologic expertise. The recommendations regarding phlebotomy therapy and anti-aggregative therapy are essentially the same as those above. Aspirin appears to decrease the rate of spot anabortions and pregnancy complications [50]. Due to lack of evidence for teratogenicity or influence on live birth rate [51] with IFNα therapy is generally possible and especially recommended in high-risk pregnancies [52].
Take-Home Messages
- Polycythemia vera is a myeloproliferative neoplasia of hematopoietic stem cells predominantly triggered by somatic mutations of the JAK2 gene and resulting clonal autonomous hematopoiesis.
- Clinically, mirco- and macrovascular symptoms and complications in the setting of erythrocytosis and frequent concomitant thrombocytosis are prominent. Of further prognostic relevance is the risk of myelofibrosis as well as leukemic transformation.
- In addition to clinical and laboratory parameters, the diagnostic algorithm primarily involves the detection of a JAK2 mutation in the bone marrow, with other genetic aberrations becoming increasingly important.
- Therapeutically, patients are divided into risk groups. While phlebotomy therapy and platelet aggregation inhibitors are used regardless of risk, cytoreductive therapy is indicated primarily in high-risk patients.
- Pregnancies in PV patients are always considered high-risk pregnancies and should be managed multidisciplinary in specialized centers.
Literature:
- Arber DA, et al: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood, 2016. 127(20): 2391-2405.
- Barbui T, et al: The 2016 revision of WHO classification of myeloproliferative neoplasms: clinical and molecular advances. Blood Rev, 2016. 30(6): 453-459.
- Jamieson CH, et al: The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci U S A, 2006. 103(16): 6224-6229.
- Bento C, et al: Genetic basis of congenital erythrocytosis: mutation update and online databases. Hum Mutat, 2014. 35(1): 15-26.
- Polycythemia vera: the natural history of 1213 patients followed for 20 years. Gruppo Italiano Studio Policitemia. Ann Intern Med, 1995. 123(9): 656-664.
- Moulard O, et al: Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol, 2014. 92(4): 289-297.
- Tefferi A, et al: Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood, 2014. 124(16): 2507-2513; quiz 2615.
- Crisa E, et al: A retrospective study on 226 polycythemia vera patients: impact of median hematocrit value on clinical outcomes and survival improvement with anti-thrombotic prophylaxis and non-alkylating drugs. Ann Hematol, 2010. 89(7): 691-699.
- Barbui T., et al: Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol, 2011. 29(23): 3179-3184.
- Tefferi A, et al: Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia, 2013. 27(9): 1874-1881.
- Koschmieder S, et al: Myeloproliferative neoplasms and inflammation: whether to target the malignant clone or the inflammatory process or both. Leukemia, 2016. 30(5): 1018-1024.
- Kroll MH, Michaelis LC, Verstovsek S: Mechanisms of thrombogenesis in polycythemia vera. Blood Rev, 2015. 29(4): 215-221.
- Finazzi G, Barbui T: Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leukemia, 2008. 22(8): 1494-1502.
- Mesa RA, et al: The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer, 2007. 109(1): 68-76.
- Saini KS, Patnaik MM, Tefferi A: Polycythemia vera-associated pruritus and its management. Eur J Clin Invest, 2010. 40(9): 828-834.
- Michiels JJ: Erythromelalgia and vascular complications in polycythemia vera. Semin Thromb Hemost, 1997. 23(5): 441-454.
- De Stefano V, et al: Splanchnic vein thrombosis and myeloproliferative neoplasms: molecular-driven diagnosis and long-term treatment. Thromb Haemost, 2016. 115(2): 240-249.
- Dentali F, et al: Cerebral venous thrombosis and myeloproliferative neoplasms: results from two large databases. Thromb Res, 2014. 134(1): 41-43.
- Elliott MA, Tefferi A: Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. Br J Haematol, 2005. 128(3): 275-290.
- Michiels JJ, et al: The paradox of platelet activation and impaired function: platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost, 2006. 32(6): 589-604.
- Pardanani A, et al: Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia, 2007. 21(9): 1960-1963.
- Vannucchi AM, et al: Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: a critical reappraisal. Leukemia, 2008. 22(7): 1299-1307.
- Passamonti F, et al: A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia, 2010. 24(9): 1574-1579.
- Ward AC, Touw I, Yoshimura A: The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood, 2000. 95(1): 19-29.
- James C, et al: A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature, 2005. 434(7037): 1144-1148.
- Baxter EJ, et al: Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet, 2005. 365(9464): 1054-1061.
- Gangat N, et al: Cytogenetic studies at diagnosis in polycythemia vera: clinical and JAK2V617F allele burden correlates. Eur J Haematol, 2008. 80(3): 197-200.
- Tefferi A, et al: Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv, 2016. 1(1): 21-30.
- Scherber R, et al: The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood, 2011. 118(2): 401-408.
- Grinfeld J, et al: Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N Engl J Med, 2018. 379(15): 1416-1430.
- Marchioli R, et al: Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med, 2013. 368(1): 22-33.
- Landolfi R, et al: Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med, 2004. 350(2): 114-124.
- Alvarez-Larran A, et al: Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood, 2010. 116(8): 1205-1210; quiz 1387.
- Grunwald MR, et al: Clinical and Disease Characteristics From REVEAL at Time of Enrollment (Baseline): Prospective Observational Study of Patients With Polycythemia Vera in the United States. Clin Lymphoma Myeloma Leuk, 2018. 18(12): 788-795 e2.
- Michiels JJ, et al: Platelet-mediated erythromelalgic, cerebral, ocular and coronary.
InFo ONCOLOGY & HEMATOLOGY 2019; 7(2-3): 12-15.









