Systemic vasculitides are inflammatory syndromes of the blood vessels, which can cause a very broad spectrum of symptoms depending on the affected vessel caliber and localization. The most common primary vasculitides in adults are described with current drug therapy options.
Systemic vasculitides are inflammatory syndromes of the blood vessels, which can cause a very broad spectrum of symptoms depending on the affected vessel caliber and localization. Vasculitic involvement of small vessels, i.e., capillaries, arterioles, and venules, may manifest, for example, as palpable purpura of the skin, rapid-progressive necrotizing glomerulonephritis with renal failure, pulmonary hemorrhage, nosebleeds, scleritis, or cerebral opacification, depending on the localization. If medium-sized and large arteries are affected, then there is a risk of tissue infarcts, aneurysms, bleeding and thrombosis. Although significant progress has been made in the treatment of systemic vasculitides over the past 40 years, mortality remains markedly increased compared with the general population [1]. The most common primary vasculitides in adults are described below, along with current drug therapy options.
Classification of systemic vasculitides
The group of “primary” systemic vasculitides comprises independent “idiopathic” disease syndromes, whereas “secondary” vasculitides occur in association with pre-existing diseases. Examples of secondary vasculitides include hepatitis C-associated cryoglobulinemic vasculitis and vasculitis associated with long-standing seropositive rheumatoid arthritis.
The classification of primary systemic vasculitides according to the Chapel-Hill classification is based on the caliber of vessel affected [2] (Overview1). Large-vessel vasculitides affecting the aorta and/or its branches include Takayasu arteritis, which occurs mainly in young Asian women, and giant cell arteritis (RZA; synonym: temporal arteritis), which usually does not manifest before the age of 50. Kawasaki syndrome, which affects children, and the very rare panarteritis nodosa affect medium-sized arteries in particular. Primary small vessel vasculitides fall into two main groups: ANCA (anti-neutrophil cytoplasmic antibodies)-associated vasculitides (AAV) and ANCA-negative vasculitides. AAVs include granulomatosis with polyangiitis (GPA; formerly Wegener’s disease); microscopic polyangiitis (MPA); eosinophilic granulomatosis with polyangiitis (EGPA; formerly Churg-Strauss syndrome); and focal necrotizing glomerulonephritis (FNGN). ANCA-negative primary small vessel vasculitides are associated with a number of immune complex-mediated vasculitides, e.g. IgA vasculitis (Henoch-Schönlein), which occurs mainly in children, and essential cryoglobulinemic vasculitis.

Secondary vasculitides usually involve small vessels (capillaries, arterioles, venules), and like ANCA-negative small vessel vasculitides, they are usually caused by immune complexes with activation of the complement system. Treatment of secondary vasculitides is always in relation to the underlying disease and, given the benefit-risk constellation, very often involves the short-term use of higher-dose glucocorticoids, and in some cases (e.g., viral-induced cryoglobulinemic vasculitis), therapeutic depletion of CD20+ B cells [3]. Behçet’s disease, in which all vascular calibers can be affected, occupies a certain special position among the vasculitis syndromes.
In all vasculitides, the first treatment goal is to achieve as rapid a clinical remission as possible, which is referred to as remission induction. This is followed by the remission-maintenance phase, which can last several years depending on the type of vasculitis.
Giant cell arteritis (RZA)
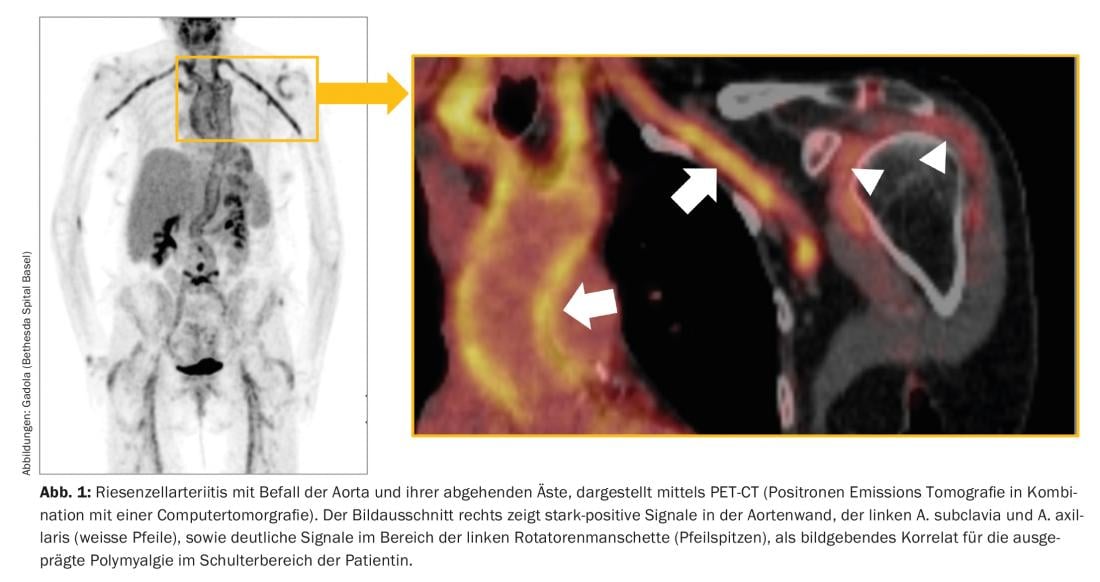
RZA, also known as arteritis temporalis, is the most common vasculitis in our latitudes and often occurs after the age of 60, and in the vast majority of cases after the age of 50. It affects the aorta and the large and medium-sized arteries branching off from it (Fig. 1). Typical symptoms are new, often “neuralgic” headaches, a polymyalgic syndrome, B-symptomatics with a drop in performance, exhaustion and night sweats, claudication symptoms of the extremities, tongue and/or masticatory muscles, and visual disturbances. RZA is feared mainly because of the risk of acute blindness, which is usually irreversible. Laboratory evidence suggests a pronounced acute-phase reaction is typical – but not obligatory – with markedly accelerated sedimentation response (BSR) and elevation of C-reactive protein (CRP).
Glucocorticoids are the first-line medications for RZA and typically result in dramatic improvement of subjective symptoms within 24 hours. If ocular involvement is suspected, e.g., transient disturbances of visual acuity, and potential threat of blindness, high doses, e.g., 1 g methylprednisolone for three consecutive days, are given intravenously. In other cases, an initial daily dose of 40-60 mg is sufficient. The dose is adjusted according to the clinic and laboratory values. Until the prednisone dose can be reduced below 20 mg/day, antibiotic prophylaxis with sulfometoxazole-trimetoprim (e.g., Cotrim-CT 800/160 mg; Bactrim forte®) 3×1/week should be given to prevent opportunistic infections, especially those caused by Pneumocystis jirovecii.
Previously, prednisone doses of 1 mg/kg body weight were used for a full year in RZA, as in other vasculitides, with frequent occurrence of severe side effects. However, with the help of steroid-sparing basic anti-rheumatic drugs (disease-modifying anti-rheumatic drugs, DMARDs), the prednisone dose can be reduced to below the so-called steroid threshold dose of 7.5 mg prednisone much earlier. Methotrexate (MTX) in particular has been shown to be an effective DMARD for RZA. The onset of action is delayed with MTX, as with all DMARDs, and occurs approximately 4-6 weeks after the effective dose is reached.
Methotrexate should always be administered parenterally, i.e. subcutaneously (s.c.) once a week, for the treatment of vasculitides. If well tolerated, the dose may be increased to a maximum of 0.3 mg/kgKG per week. BSR and CRP also serve as valuable progression biomarkers of inflammatory activity under treatment with prednisone and DMARDs. To reduce toxic side effects of MTX, folic acid, e.g., 10 mg/week, must always be taken concomitantly the day after the methotrexate injection. MTX must never be combined with sulfometoxazole-trimetoprim or severe myelosuppression may occur. Therefore, we use MTX only after the dose of prednisone has been reduced to less than 20 mg/day.
Prior to initiation of steroid-sparing basic therapy with MTX or other DMARDs, a chest radiograph should be obtained to exclude chronic infection or pulmonary fibrosis, a differential blood count, liver and kidney enzymes, and serologic testing for hepatitis B, hepatitis C, and HIV. During the first 3 months under methotrexate, monthly checks of liver and kidney values and blood count are recommended; thereafter, these checks may be performed at longer intervals of 8 to 12 weeks, if necessary. Detailed recommendations for the use of DMARDs in rheumatologic diseases can be found on the Internet portals of the rheumatologic societies [e.g., 4].
Since 2017 (EU area) resp. 2018 (Switzerland), the anti-interleukin-6 receptor (anti-IL6R) antibody tocilizumab (Actemra®) is approved for the treatment of RZA. With tocilizumab, the glucocorticoid dose can be reduced much more rapidly, even without methotrexate, with good clinical outcome [5,6]. Tocilizumab therapy should be given for at least 1 year, otherwise recurrences are common [7]. BSR and CRP are suppressed under tocilizumab and therefore not useful as progression parameters of disease activity. Recent data suggest that the glucocorticoid dose under tocilizumab may be discontinued very rapidly, i.e., within a few weeks [8]; however, further studies are needed before clear recommendations can be made. Since glucocorticoids in RZA cannot currently be lowered below the threshold dose for a prolonged period (>3 months), the rapid initiation of antiresorptive therapy, e.g., with alendronate, is recommended to prevent steroid-acquired bone resorption, resp. of osteoporosis.
Takayasu arteritis (TA)
Similar to RZA, TA affects the aorta and large arteries branching from it. Unlike RZA, this vasculitis occurs in 80-90% of cases in women, with onset between the ages of 10 and 40 . TA progresses in episodes, and typically manifests with constitutional symptoms, arthralgias, and – characteristically – marked carotid pressure pain (in 10-30%). During the course of the disease, vascular occlusion, severe renovascular hypertension, (Takayasu) retinopathy, and aortic aneurysms with or without aortic valve regurgitation may occur.
When TA is diagnosed, glucocorticoids are used in the first instance. Steroid-sparing disease-modifying DMARDs most commonly used in TA are methotrexate s.c. (as in RZA) or azathioprine p.o. (up to 2 mg/kgKG). Alternatives are mycophenolate (1.5 g-3 g/day p.o.) and leflunomide (20 mg/day p.o.). In cases of intolerance to methotrexate or oral DMARDs, treatment-resistant and severe cases, TNFalpha- blockers (e.g. etanercept or infliximab) or other biologics (e.g. tocilizumab, abatacept, ustekinu-mab) are used, but these are not yet approved for this indication [9].
ANCA-associated vasculitides (AAV)
GPA (formerly Wegener’s disease)
GPA is the most common primary small vessel vasculitis in our latitudes, with an almost balanced gender distribution and typical age of manifestation between 40-60 years of age. Year of life. A distinction is made between a non-vasculitic, granulomatous “initial phase” (localized) and a systemic, vasculitic “generalization phase”, which may occur sequentially (initial phase ‘ generalization phase) or simultaneously. While a wide range of effective therapies is now available for the treatment of small vessel vasculitis, the treatment of aggressive granulomatous inflammatory manifestations is often a major challenge [10,11].
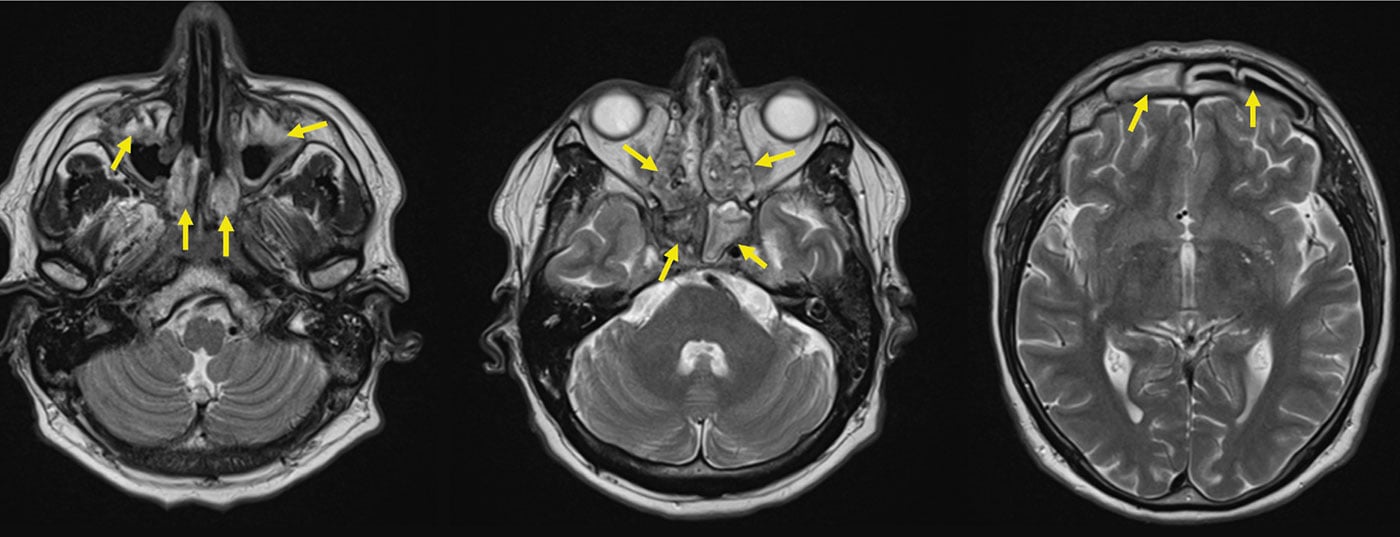
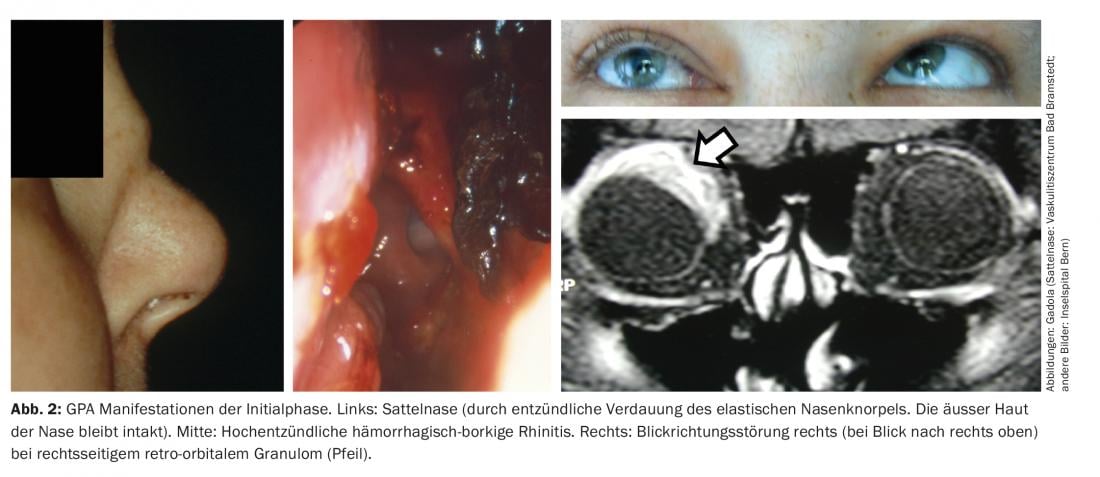
Initial phase of the GPA (“localized” GPA). The initial phase of GPA typically manifests in the ear, nose, throat, and airways, e.g., in the form of chronic hemorrhagic borky rhinitis, refractory sinusitis, or mastoiditis. Aggressive progression may result in a saddle nose, due to destruction of the elastic nasal cartilage, fistula formation into the orbit or toward the face, retro-orbital granulomas with impaired direction of gaze (diplopia), and granulomatous pachymeningitis (Fig. 2). The latter two probably as a result of granulomatous inflammation originating from the sinuses per continuitatem [11]. ANCA with specificity for proteinase-3 (PR3-ANCA) or, more rarely, myeloperoxidase (MPO-ANCA) are detectable during the initial phase in only about 50% of GPA patients.
Various drug combinations are used to treat the “pure” initial phase without concomitant systemic vasculitis, depending on the severity of symptoms. In milder cases, sulfomethoxazole-trimetoprim (T/S) is used with or without low-dose prednisone (10 mg/day). The use of T/S dates back to an empirical observation in the 1970s by Richard Deremee at the Mayo Clinic [12]. In the 1990s, an association between chronic nasal colonization by Staphylococcus aureus and disease activity in GPA was demonstrated [13]. Since then, topical intra-nasal antibiotic treatment with mupirocin has also been used, but without resounding success. A recent analysis of the endonasal microbiome in GPA has shown an interesting association of disease activity in GPA with Corynebacterium tuberculostearicum [14]. This pathogen is an important pathogen in other granulomatous diseases [15]. Corynebacterium tuberculostearicum is resistant to most antibiotics [16], which may explain the moderate success of T/S and mupirocin.
If the initial phase is more aggressive, methotrexate (always without T/S, otherwise combined bone marrow toxicity) is primarily used in combination with prednisone. In refractory cases, large lung granulomas and also retro-orbital granulomas, therapy with anti-CD20 antibodies (rituximab) may be effective [17]. The granulomas in GPA have a high density of CD20+ B cells and are seen as a possible site of origin of ANCA production [18,19]. A particular manifestation of GPA during the initial phase is inflammatory fibrosing tracheal stenosis with dyspnea and inspiratory stridor, which may require treatment with local glucocorticoid infiltration and balloon dilatation.
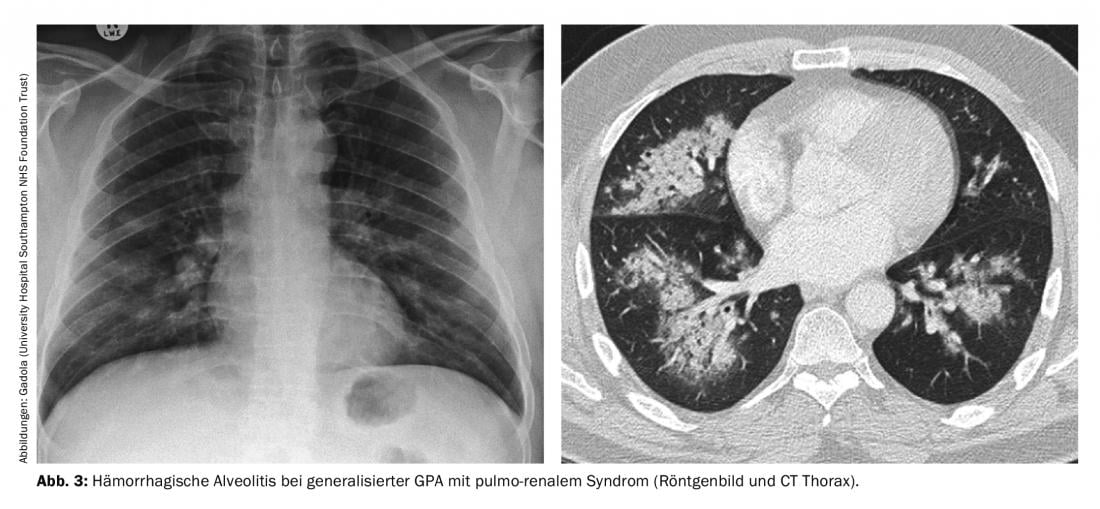
Generalisaton phase of GPA (ANCA in >98%). Systemic small vessel vasculitis in GPA, in which PR3-ANCA or MPO-ANCA can virtually always be detected in serum, can affect all organs. Infection of the kidneys typically leads to rapid-progressive, necrotizing, “pauci-immune” glomerulonephritis (RPGN), which, if untreated, can rapidly lead to severe renal failure requiring dialysis. Particularly feared is the pulmo-renal syndrome, resp. The combined occurrence of RPGN with hemorrhagic alveolitis (Fig. 3), which has a high mortality. Other typical manifestations of the generalization phase include marked B-symptomatology, scleritis-typically painful nodular scleritis-which can lead to scleromalacia, often very painful mononeuritis multiplex, encephalitis with opacification, palpable purpura, and nonerosive polyarthritis. The following treatment principles for generalized GPA can be applied to MPA and FNGN, and to some extent to EGPA.
Remission induction in generalized GPA. Different modes of action are available to induce remission, depending on the severity of the manifestation. Primarily, in generalized GPA, inflammatory activity should always be controlled as quickly and thoroughly as possible.
In the case of non-renal and also otherwise not organ-threatening systemic GPA, remission induction therapy can be started with methotrexate plus prednisone or with intravenous (i.v.) cyclophosphamide pulses (e.g. 4 pulses of 10 mg/kgKG each at 3-week intervals) plus prednisone, provided that close clinical progress monitoring at least once a week is possible. For renal manifestations and other organ-threatening manifestations, in addition to prednisone, primary use is cyclophosphamide orally (starting at 2 mg/kgKG/day) or as an i.v. pulse, or rituximab (2 i.v. infusions of 1g rituximab each 14 days apart) in combination with prednisone. The choice between the more dose-intensive oral cyclophosphamide treatment and pulse therapy always requires careful consideration of the risk-benefit ratio by the specialist experienced with it. Under cyclophosphamide, leukocytes in particular, and especially neutrophil granulocytes, which are typically markedly elevated in active GPA vasculitis, must be determined regularly. Leukocytes should reach nadir within 8-10 days on cyclophosphamide, which should always be determined. If leukocytes do not fall, then this indicates continued disease activity, which may necessitate “leukocyte-adapted” dose increases to 2.5-3 mg/kgKG. Under cyclophosphamide, severe opportunistic infections may occur shortly after initiation of therapy, when leukopenia occurs, particularly reactivation of cytomegalovirus (CMV). Typical dose-related toxic bladder side effects include hemorrhagic cystitis and bladder carcinomas, which occur after a cumulative dose of at least 25 g (and an average of 100 g) of cyclophosphamide [20], and myelodysplastic syndrome (MDS). The likelihood of bladder complications can be reduced by good hydration throughout the treatment period and the additional use of Mesna (Uromitexan), which binds and neutralizes the cyclophosphamide metabolite acrolein.
Long-lasting B-cell depletion with secondary humoral immunodeficiency and frequent severe respiratory infections may occur with rituximab, especially in patients who have received cyclophosphamide previously [21].
In particularly severe and/or refractory courses of systemic GPA, additional measures and medications are sometimes used, including plasmapheresis, intravenous immunoglobulin administration, mycophenolate mofetil (MMF), the B- and T-cell-depleting anti-CD52 antibody alemtuzumab, anti-thymocyte globulin, 15-deoxyspergualin – an inhibitor of cell differentiation – and hematopoietic stem cell transplantation. The success of these intensified treatments is not well established, and their use should be exclusive to experienced centers.
A very promising new mechanism of action for the treatment of generalized GPA as well as other AAV is inhibition of the complement system. Avacopan, an oral antagonist of the C5a receptor, has shown very promising results in Phase 3 trials, so the likelihood of approval in the near future is high. In patients with generalized GPA who received Avacopan in addition to standard therapy (with cyclophosphamide or rituximab), glucocorticoids were discontinued very rapidly. Interestingly, renal function in Avacopan-treated patients showed continuous improvement throughout the 52-week treatment period [22].
An existing drug to inhibit the complement system is the anti-C5 antibody eculizumab (Soliris®), which has been used sporadically for aggressive progression of AAV [e.g., 24]. Unfortunately, a phase 2 trial of eculizumab in ANCA vasculitis was withdrawn before patient enrollment (NCT01275287). Another drug, iptacopan (LNP023), which inhibits efficient activation of the complement system via factor B, has recently received approval for the treatment of C3 glomerulopathy (C3G), and it is quite possible that this drug will be used in AAV in the future.
Remission maintenance in generalized GPA. After clinical remission has been achieved, remission-maintaining therapy is intended to safeguard the inflammatory activity of AAV even with small doses of prednisone or even without concomitant glucocorticoids. Since 1995, the European Vasculitis Study Group (EUVAS) has conducted a large number of clinical intervention studies in AAV. MTX, azathioprine, or rituximab are recommended to maintain remission in GPA and other AAV. Rituximab may be the most effective drug of the three, although the optimal dosage and dose interval of rituximab in AAV is still under debate. Some experts recommend fixed 6-month doses of 1g rituximab [24], while a French comparative study of rituximab versus azathioprine showed good results for a 6-12 month interval with 500mg doses [25]. With rituximab, as with cyclophosphamide, PR3 or MPO-ANCA levels fall below the detection limit in most cases, correlating with clinical remission of vasculitis. In these patients, in our experience, the rituximab interval can be adjusted to match the rebound in ANCA titers, extending the interval to more than 12 months in some cases.
Other AAV (MPA, FNGN, EGPA)
MPA and FNGN
In contrast to GPA, granulomas do not occur in MPA and FNGN. These AAV are slightly more commonly associated with MPO-ANCA than PR3-ANCA, but this does not matter for treatment. Both AAV typically manifest as “pauci-immune” rapid progressive glomerulonephritis (RPGN). The term “pauci-immune” refers to the weak to absent detectability of immunoglobulin deposits in the immunohistochemical examination of renal biopsies. Other typical manifestations in MPA include painful mononeuritis multiplex and alveolitis, which can lead to pulmonary fibrosis with significantly increased mortality in approximately one-third of patients [26]. Treatment of MPA and FNGN is analogous to the treatment principles described above for generalized GPA.
EGPA (formerly: Churg-Strauss syndrome/vasculitis)
The EGPA can be described as the “atopic counterpart” to the GPA. It is seen in patients with years to decades of pre-existing atopic symptoms, particularly asthma and/or chronic pansinusitis (Fig. 4) with nasal polyps. During the initial phase of EGPA, granulomas are found in the affected tissue, similar to GPA. In contrast to GPA, however, these granulomas are not densely interspersed with neutrophil granulocytes but with eosinophil granulocytes. In the peripheral blood of EGPA patients, eosinophilia is found analogously. EGPA differs clinically from GPA in important ways. The occurrence of RPGN is less frequent than in GPA, approximately 15-20% [27], but, as in GPA, is strictly associated with the presence of ANCA. Neurologic manifestations, especially often very painful mononeuritis multiplex [28], occur in more than half of patients. What is feared in EGPA are the cardiac manifestations, e.g., vasculitis of the coronary arteries, which occur in approximately 40% of patients and are the most common cause of death in EGPA [29,30].
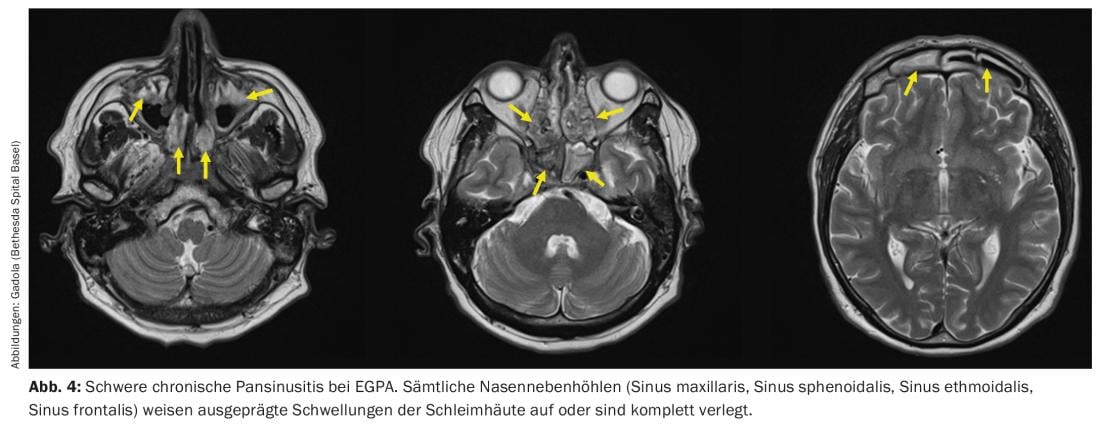
The therapy of EGPA is very similar to that of generalized GPA, and the most common drugs in EGPA, as in GPA, are glucocorticoids, MTX, cyclophosphamide, and rituximab. High-dose glucocorticoids very rapidly cause a dramatic decrease in eosinophils and clinical improvement. However, as with GPA, steroid therapy should be reduced rapidly and to the prednisone threshold dose within 3 months to reduce the risk of severe airway infections. For relapsed or refractory EGPA, additional interferon-alpha (3×/week to daily administration s.c.) and the anti-IL5 antibody mepolizumab (Nucala®, 300 mg s.c. every 4 weeks) are available, which is also approved for this indication. With effective therapy, the eosinophilia should disappear and the ANCA titer should decrease.
Non-ANCA-associated small vessel vasculitis.
Essential cryoglobulinemic vasculitis.
This small vessel vasculitis is caused by in situ activation of the complement cascade in vessel walls after deposition of type II (less commonly type III) cryoglobulins. It manifests primarily on the skin with urticarial vasculitis or palpable purpura (Fig. 5) . In renal involvement, lupus-like immune complex and complement-mediated membranoproliferative glomerulonephritis is usually found. Other typical symptoms are myalgias and arthralgias, as well as polyneuropathy. During relapses, the complements C4 (“always”) and C3 (“frequently”) are decreased in the serum.
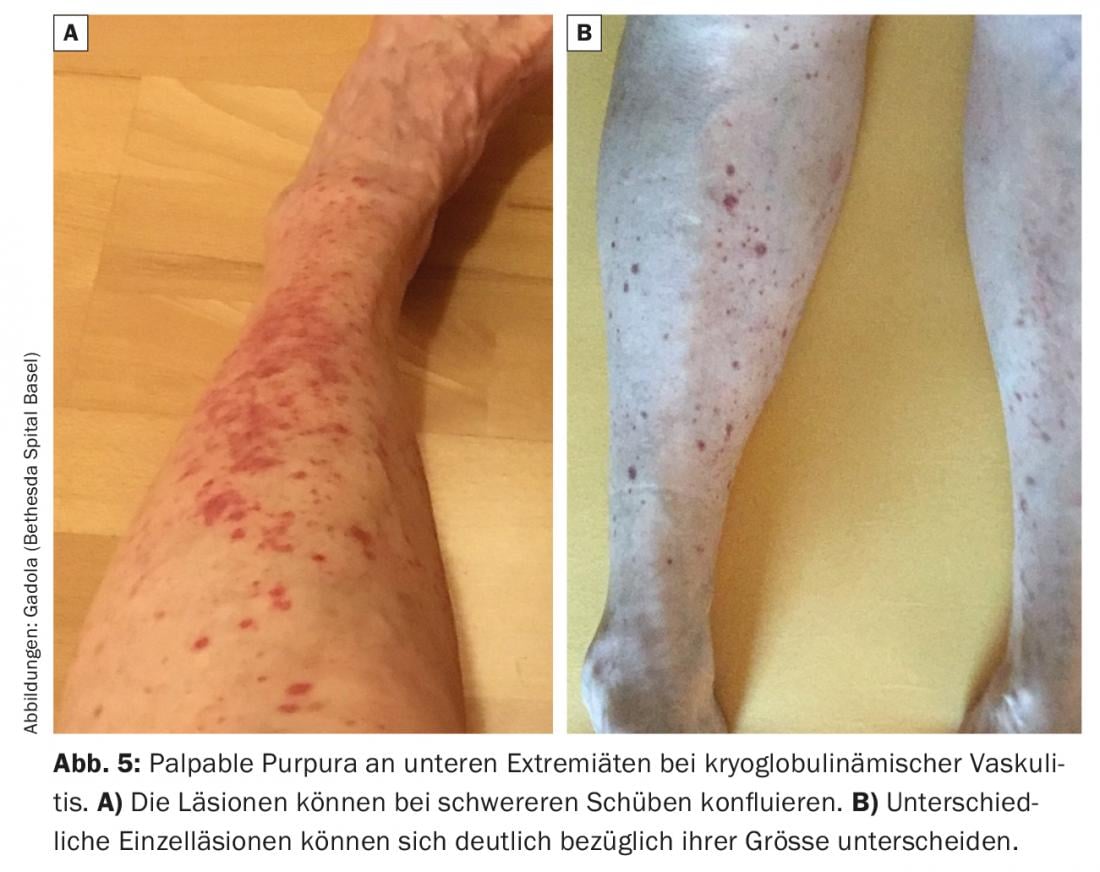
Therapy begins with glucocorticoids and is supplemented with DMARDs (MTX or azathioprine) as needed. In refractory cases, rituximab has been shown to be very effective, and in severe acute cases with severe renal involvement, cyclophosphamide is used [31].
More common than essential cryoglobulinemic vasculitis, in which the etiology is by definition unknown, are HCV-associated cryoglobulinemic vasculitis (type II or type III cryoglobulins in serum) and cryoglobulinemic vasculitis episodes in B-cell neoplasms (type I or type III cryoglobulins). Here, the primary focus is on treatment of the underlying disease, as well as B-cell depleting treatment with rituximab in severe cases.
Leukocytoclastic vasculitis
Usually, immune complex-mediated leukocytoclastic vasculitis is strictly confined to the capillaries and venules of the dermis and manifests with pruritic to painful palpable purpura of the lower extremities. If other organs are affected besides the skin, another primary or secondary small vessel vasculitis must always be sought. Leukocytoclastic vasculitis confined to the skin. is often self-limiting; however, severe recurrent or chronic courses may occur, which are treated with glucocorticoids and, if necessary, with various DMARDs; in particularly severe cases, also with cyclophosphamide.
Other “idiopathic” (and thus “primary”) small-vessel vasculitides of the adult include anti-C1q antibody-associated hypocomplementemic urticarial vasculitis (HUV), IgA vasculitis (Henoch-Schönlein), and anti-GBM (GBM, glomerular basement membrane) vasculitis. The therapy of these vasculitides is largely similar to the principles described above for other small-vessel vasculitides.
Behçet’s disease
Behçet’s disease is characterized by a broad clinical spectrum and a relapsing course. Vasculitis in Behçet’s disease may involve arterial and/or venous vessels of all caliber sizes. Pulmonary artery vasculitis with consecutive aneurysm formation and rupture into lung tissue is the most common direct cause of death. Severe complications are also caused by inflammatory venous thrombosis, cardiac and cerebral involvement.
Behçet’s disease has some striking clinical similarities to Crohn’s disease, such as the presence of colitis, enterocolitic and perianal fistula formations, mucocutaneous aphthae, arthitides, erythema nodosum, and uveitides. The therapy of gastrointestinal manifestations in Behçet’s disease reflects this: in addition to glucocorticoids, 5-aminosalycylic acid (5-ASA), azathioprine, and, in case of insufficient response, TNFalpha blockers are used. Depending on the severity, topical steroids, colchicine or the phosphodiesterase-4 inhibitor apremilast (Otezla®), which has been approved for this purpose, can be used for the treatment of mucocutaneous aphthae. In acute ocular involvement, systemic glucocorticoids (e.g., 1g methylprednisolone i.v. for hypopyon uveitis) are primarily recommended, always in combination with a DMARD, most notably cyclosporine A or azathioprine, or with interferon-alpha or a TNFalpha blocker (infliximab or adalimumab) [32]. In cases of vasculitic involvement of the pulmonary arteries, high-dose glucocorticoids in combination with cyclophosphamide or TNFalpha blockers are effective. Non-drug interventions are also used in severe Behçet’s disease. When bleeding from a large pulmonary artery aneurysm is imminent, embolization treatment is primarily recommended rather than open thoracic surgical vascular revision. In cases of severe gastrointestinal bleeding, impending intestinal perforation or intestinal strictures, patients require emergency surgery.
Take-Home Messages
- In all vasculitides, the first treatment goal is to achieve as rapid a clinical remission as possible. This is followed by the remission-maintenance phase, which can last for several years.
- MTX must never be combined with sulfometoxazole-trimetoprim in the treatment of RZA, or severe myelosuppression may occur.
- A promising new mechanism of action for the treatment of generalized GPA as well as other AAV is inhibition of the complement system.
- Systemic small vessel vasculitis in GPA can affect all organs. Infection of the kidneys typically leads to rapid progressive, necrotizing, “pauci-immune” glomerulonephritis (RPGN), which can lead to severe renal failure if left untreated.
Literature:
- Jardel S, et al: Mortality in systemic necrotizing vasculitides: A retrospective analysis of the French Vasculitis Study Group registry. Autoimmune Rev 2018 Jul; 17(7): 653-659.
- Jennette JC, Falk RJ, Bacon PA, et al: 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013 Jan; 65(1): 1-11.
- Chung L, et al: Successful Use of Rituximab for Cutaneous Vasculitis. Arch Dermatol 2006; 142(11): 1407-1410; doi:10.1001/archderm.142.11.1407.
- www.rheuma-net.ch/de/fachinformationen/behandlungsempfehlungen
- Villiger PM, Adler S, Kuchen S, et al: Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016; 387: 1921-1927.
- Stone JH, Tuckwell K, Dimonaco S et al: Trial of tocilizumab in giant-cell arteritis. N Engl J Med 2017; 377: 317-328.
- Christ L, et al: A Proof of Concept Study to Assess the Efficacy of Tocilizumab in Combination with Ultra-Short Glucocorticoid Administration to Treat Newly Diagnosed Giant Cell Arteritis – a 24 Week Analysis. Arthritis Rheumatol 2020; 72 (suppl 10).
- Adler S, et al: Risk of relapse after discontinuation of tocilizumab therapy in giant cell arteritis. Rheumatology (Oxford) 2019 Sep 1; 58(9): 1639-1643.
- Hellmich B, et al: 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2020 Jan; 79(1): 19-30.
- Gadola SD, Gross WL: The renaissance of granulomatous inflammation in AAV. Nat Rev Rheumatol 2012 Jan 10; 8(2): 74-76.
- Holle JU, et al: Orbital masses in granulomatosis with polyangiitis are associated with a refractory course and a high burden of local damage. Rheumatology 2013; 52: 875882.
- DeRemee RA, et al: Wegener’s granulomatosis: observations on treatment with antimicrobial agents. Case Reports Mayo Clin Proc 1985 Jan; 60(1): 27-32; doi: 10.1016/s0025-6196(12)65279-3.
- Stegeman CA, et al: Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med 1994 Jan 1; 120(1): 12-17.
- Rennie Rhee D, et al: Nasal Bacteria Associated with Disease Activity and ANCA Levels in Granulomatosis with Polyangiitis. Arthritis Rheumatol 2020; 72 (suppl 10).
- Taylor GB, et al: A clinicopathological review of 34 cases of inflammatory breast disease showing an association between corynebacteria infection and granulomatous mastitis. Pathology 2003 Apr; 35(2): 109-119.
- Dobinson HC, et al: Antimicrobial Treatment Options for Granulomatous Mastitis Caused by Corynebacterium Species. J Clin Microbiol 2015 Sep; 53(9): 2895-2899.
- Holle JU, Dubrau C, Herlyn K, et al: Rituximab for refractory granulomatosis with polyangiitis (Wegener’s granulomatosis): comparison of efficacy in granulomatous versus vasculitic manifestations. Ann Rheum Dis 2012; 71: 327-333.
- Voswinkel J, Mueller A, Kraemer JA, et al: B lymphocyte maturation in Wegener’s granulomatosis: a comparative analysis of VH genes from endonasal lesions, Ann Rheum Dis 2006; 65: 859-864.
- Voswinkel J, Assmann G, Held G et al: Single cell analysis of B lymphocytes from Wegener’s granulomatosis: B cell receptors display affinity maturation within the granulomatous lesions. Clin Exp Immunol 2008; 154: 339-345.
- Knight A, et al: Urinary bladder cancer in Wegener’s granulomatosis: risks and relation to cyclophosphamide. Ann Rheum Dis 2004.
- Thiel J, et al: B cell repopulation kinetics after rituximab treatment in ANCA-associated vasculitides compared to rheumatoid arthritis, and connective tissue diseases: a longitudinal observational study on 120 patients. Arthritis Res Ther 2017 May 18; 19(1): 101.
- Merkel P, et al: The Effect on Renal Function of the Complement C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. Arthritis Rheumatol 2020; 72 (suppl 10).
- Huizenga N, et al: Treatment of Aggressive Antineutrophil Cytoplasmic Antibody-Associated Vasculitis With Eculizumab. Kidney Int Rep 2020 Apr; 5(4): 542-545.
- Smtih R, et al: Extended Follow-Up of Patients Recruited to a Randomized, Controlled Trial of Rituximab versus Azathioprine After Induction of Remission with Rituximab for Patients with ANCA-Associated Vasculitis and Relapsing Disease. Arthritis Rheumatol 2020; 72 (suppl 10).
- Guillevin L, et al: Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med 2014 Nov 6; 371(19): 1771-1780.
- Tzelepis GE, et al: Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. European Respiratory Journal 2010; 36: 116-121.
- Sinico RA, et al: Renal involvement in Churg-Strauss syndrome. Am J Kidney Dis 2006 May; 47(5): 770-779.
- Wolf J, et al: Neurologic complications of Churg-Strauss syndrome: a prospective monocentric study. Current Neurology 2009; 36: V188.
- Conron M, Beynon HL: Churg-Strauss syndrome. Thorax 2000 Oct; 55(10): 870-877.
- Solans R, et al: Churg-Strauss syndrome: outcome and long-term follow-up of 32 patients. Rheumatology (Oxford) 2001; 40: 763-771.
- Braun G, et al: Cryoglobulinaemic vasculitis: classification and clinical and therapeutic aspects. Postgrad Med J 2007 Feb; 83(976): 87-94; doi: 10.1136/pgmj.2006.046078.
- Bettiol A, et al: Treating the Different Phenotypes of Behçet’s Syndrome. Front Immunol 2019 Dec 6; 10: 2830: doi: 10.3389/fimmu.2019.02830. eCollection 2019.
InFo PAIN & GERIATURE 2020; 2(2): 12-19.









