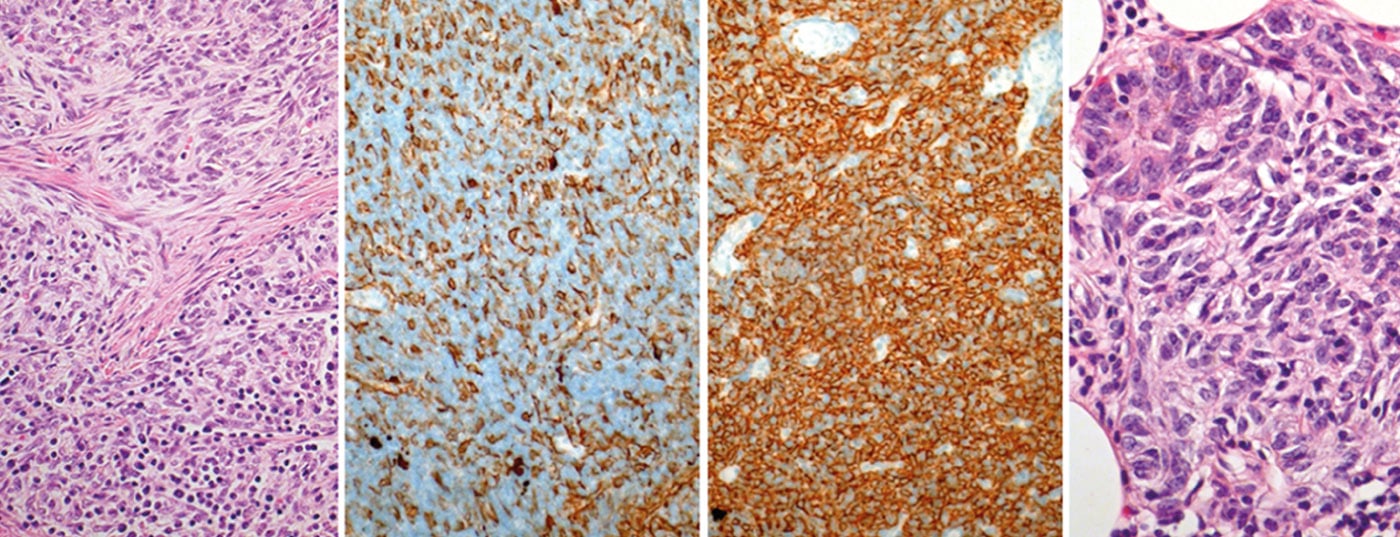
Thymic tumors are rare and belong to the so-called “orphan malignancies”. The annual incidence is approximately 0.15 per 100,000 persons. The latest classification distinguishes three major groups: Thymomas, thymic carcinomas and neuroendocrine tumors of the thymus. Due to its rarity, most knowledge on diagnosis and therapy comes from case series and monocentric studies. It was not until 2010 that an international and interdisciplinary society (International Thymic Malignancy Interest Group, ITMIG) was established to promote worldwide collaboration and initiate clinical and basic science research on thymic tumors.
Thymic tumors are rare: Their incidence is 0.15/100,000/year [1]. They can occur at any age, but show a clustering at age 30-40 years for thymomas with manifest myasthenia gravis resp. From 60-70 years of age without myasthenia [2]. Women and men are affected about equally often. About one third of patients with thymoma are asymptomatic, whereas about two thirds of patients with thymic carcinoma or a neuroendocrine tumor of the thymus present predominantly with local symptoms.
Clinical presentation
The most common symptoms, such as chest pain, cough, and shortness of breath, are caused by local growth. In invasive tumors, there may also be superior vena cava syndrome or paralysis of the phrenic nerve with diaphragmatic elevation or hoarseness due to constriction of the recurrent laryngeal nerve sinister. In addition, thymic tumors are frequently associated with paraneoplastic syndromes (Table 1).

Myasthenia gravis is by far the most common paraneoplastic syndrome. Clinical series report that approximately 30-50% of all patients with thymoma, especially type B2 thymoma, have myasthenia. Conversely, thymoma may be detected in 10-15% of patients with myasthenia. About 5% of patients with thymoma-associated myasthenia have other paraneoplastic syndromes, for example, autoimmune thyroiditis, pure red cell aplasia, limbic encephalopathy, etc. Myasthenia gravis is much rarer in thymic carcinomas – it is observed practically exclusively in carcinomas with a thymoma component. Neuroendocrine tumors of the thymus are occasionally associated with endocrinopathies.
Diagnostics
The diagnosis of thymic tumors is based on the clinical characteristics, imaging techniques and histopathological workup of biopsies resp. Resects or frozen sections. Imaging focuses on computed tomography (CT) as a modality for initial assessment and follow-up [3]. MRI can be used to evaluate suspected invasion of the heart and large blood vessels [4]. PET allows to confirm the suspected diagnosis of thymic hyperplasia versus thymoma resp. of a less aggressive tumor versus a highly aggressive tumor. Preoperative histologic confirmation has become somewhat less important, as the newer radiologic methods now have higher diagnostic reliability and complete resection of the corresponding tumors is the goal, regardless of the exact entity. In general, biopsy should be performed when the radiologic findings are unclear and lymphoma cannot be ruled out, or when the findings are unresectable, prior to initiation of induction chemotherapy or definitive radio-chemotherapy [5].
Staging and histological classification
The most commonly used staging system for thymomas is the Masoaka classification, which was last revised in 1994 (Table 2) [6].

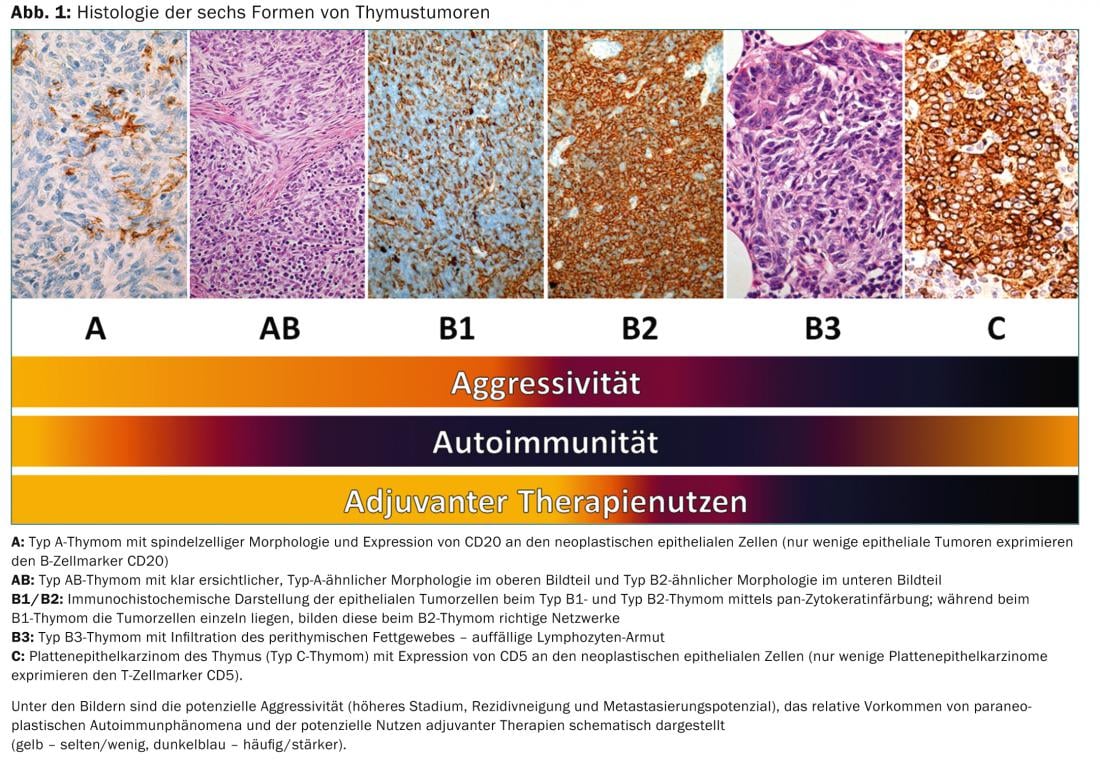
In the coming years, a TNM-based staging system will be established. There used to be different classifications for histological classification. In 1999, a histological classification based on lymphocyte/epithelial cell morphology and ratio was introduced by WHO. This classification was revised in 2004 (Tab. 3) .
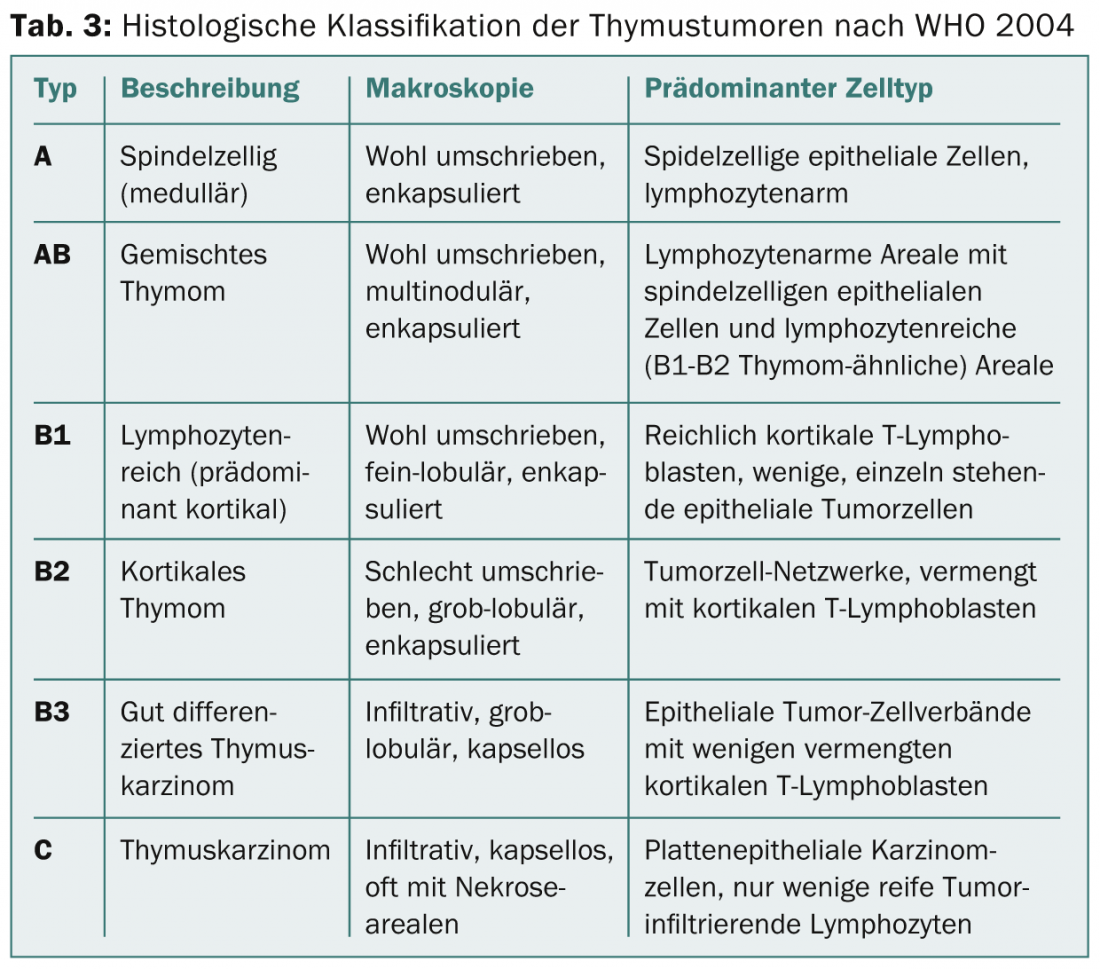
On the one hand, it reflects the tumor biology of thymic tumors (e.g., different cell origin, different aggressiveness, different accumulation of paraneoplastic autoimmunity), but it also has significance for therapeutic decisions (Fig. 1).
Therapy of thymic tumors
Several series and studies have investigated potential prognostic factors in thymic tumors. The only validated prognostic factors for survival and likelihood of recurrence are (in descending order of importance) stage at diagnosis, extent of tumor resection, and histologic subtype [7,8].
Surgery
Surgical resection is the cornerstone of treatment, and complete resection should always be attempted. The International Thymic Malignancy Interest Group (ITMIG) recommends en bloc resection with complete removal of the thymus and resection of the surrounding adipose tissue because of possible macroscopically invisible adipose tissue infiltration. Survival at ten years is stage-dependent and is 90, 70, 55, and 35% for stages I, II, III, and IVa, respectively. The corresponding recurrence rates after complete resection are 3, 11, 30, and 43% [9].
Radiotherapy
Thymic tumors tend to recur locally, which is why postoperative radiotherapy is important. A total dose of 50-54 Gy is targeted for irradiation.
Higher doses (up to 60 Gy) are recommended for incomplete resection. In masoaka stages I and II, there is no proven benefit to radiotherapy [7,10]; this also appears to be true for histologic subtypes A, AB, and B1 [11]. Nevertheless, postoperative radiotherapy is performed in stage II in many centers, as shown in a recently published survey in Europe [12]. In stage III, adjuvant radiotherapy has become established and it is also recommended in international guidelines, although there is little evidence for it from clinical trials [7,13,14]. For patients who are unresectable primarily or after induction chemotherapy, definitive radiotherapy is a treatment option, with doses up to 70 Gy being targeted. The response rate is about 70% and the probability of being alive at five years is 70-80%, which is comparable to the survival rate for incomplete resection [15,16].
Chemotherapy
Chemotherapy has a place in primary treatment, in the relapse situation and in metastases [17]. For primary unresectable stage III and IV tumors after Masoaka, neoadjuvant chemotherapy or combined radio-chemotherapy is the therapy of choice. Overall, a response rate of about 70% (29-100%) can be expected. Subsequent resection is also possible completely in about 70% of cases [2]. Combination chemotherapies were investigated in all studies.
In the postoperative setting, the value of chemotherapy is less well established. Local tumor control is the most important therapeutic goal because thymomas rarely metastasize (10-15%). Therefore, postoperative radiotherapy or, if appropriate, combined radio-chemotherapy (for unresectable tumors, inoperable patients, or for incompletely resected tumors) has priority over chemotherapy alone. In the metastatic situation or in non-resectable and already irradiated recurrences, chemotherapy can be used with palliative intent. Combination chemotherapies with an anthracycline and a platinum derivative are usually used, with response rates ranging from 50 to 90% [18]. There are no proven standard therapies for platinum-refractory tumors; various monosubstances and platinum-free combinations show response rates of 20-40% [19]. Local therapy (surgery/radiotherapy) is also important for symptom control in locally advanced tumors or in the presence of metastases.
Targeted therapies
In recent years, as in other solid tumors, targeted therapies have been increasingly investigated in thymic tumors [19]. High expression of somatostatin receptors is found in about half of thymic tumors. These patients appear to benefit from targeted therapy with octreotide, with greater benefit in thymomas than in thymic carcinomas.
Overexpression of the tyrosine kinase c-KIT is present in approximately 2% of thymomas and 80% of thymic carcinomas. Activating mutations, such as those found in gastrointestinal stromal tumors (GIST), rarely occur in thymic tumors. Therapy with the multikinase inhibitor imatinib (Gleevec®), which is directed against c-KIT among others, led to disease stabilization in advanced tumors, especially those with c-KIT mutation.
Epithelial growth factor receptor (EGFR) is overexpressed in 70% of thymomas and approximately 50% of thymic carcinomas. However, activating mutations, as seen in adenocarcinoma of the lung, are rare. Therapy with EGFR tyrosine kinase inhibitors has shown only modest benefit. The EGFR antibody cetuximab is currently being investigated in a clinical trial.
As with other tumors, neoangiogenesis plays a role in thymic tumors. Antiangiogenic agents (bevacizumab, sunitinib) have been shown to induce tumor response in small studies. Insulin-like growth factor receptor 1 (IGFR-1) is overexpressed in approximately 20% of thymic tumors. Therapy with the IGFR-1 antibody cixutumumab resulted in a high rate of disease stabilization.
Take-home-messages
- Thymic tumors are rare diseases of the anterior mediastinum and are divided into thymomas with different histologic subgroups, thymic carcinomas, and neuroendocrine tumors of the thymus.
- Thymomas are often associated with paraneoplastic syndromes, including most commonly myasthenia gravis, whereas thymic carcinomas and neuroendocrine tumors of the thymus are most commonly manifested by local symptoms.
- The basis of treatment is complete surgical resection; depending on stage and possibly subtype, postoperative radiotherapy may be evaluated.
- Chemotherapy is used in locally advanced tumors (if necessary in combination with radiation) as induction therapy before planned surgery.
- Radio-chemotherapy is performed for inoperable tumors or local recurrences.
- In metastatic tumors, combination chemotherapy with an anthracycline and a platinum-containing agent can result in a high response rate.
- In the future, better molecular understanding of thymic tumors will allow targeted therapy in certain subgroups.
Literature:
- Engels EA, Pfeiffer RM: Malignant thymoma in theUnited States: demographic patterns in incidence and associations with subsequent malignancies. Int J Cancer 2003; 105(4): 546-551. doi:10.1002/ijc.11099.
- Venuta F, et al:Thymoma and thymic carcinoma.Eur J Cardiothorac Surg 2010; 37(1): 13-25. doi:10.1016/j.ejcts.2009.05.038.
- Marom EM: Advances in thymoma imaging. J Thorac Imaging 2013; 28(2): 69-80. quiz 81-83. doi:10.1097/RTI.0b013e31828609a0.
- Sadohara J, et al: Thymic epithelial tumors: comparison of CT and MR imaging findings of low-risk thymomas, high-risk thymomas, and thymic carcinomas. Eur J Radiol 2006; 60(1): 70-79. doi:10.1016/j.ejrad.2006.05.003.
- Detterbeck FC, Parsons AM: Thymic tumors. Ann Thorac Surg 2004; 77(5): 1860-1869. doi:10.1016/j.athoracsur.2003.10.001.
- Koga K, et al: A review of 79 thymomas: modification of staging system and reappraisal of conventional division into invasive and non-invasive thymoma. Pathol Int 1994; 44(5): 359-367. www.ncbi.nlm.nih.gov/pubmed/8044305. Accessed December 2, 2014.
- Kondo K, Monden Y: Therapy for thymic epithelial tumors: a clinical study of 1,320 patients from Japan. Ann Thorac Surg 2003; 76(3): 878-884; discussion 884-885. www.ncbi.nlm.nih.gov/pubmed/12963221. Accessed December 2, 2014.
- Ströbel P, et al: Tumor recurrence and survival in patients treated for thymomas and thymic squamous cell carcinomas: a retrospective analysis. J Clin Oncol 2004; 22(8): 1501-1509. doi:10.1200/JCO.2004.10.113.
- Koppitz H, et al: State-of-the-art classification and multimodality treatment of malignant thymoma. Cancer Treat Rev 2012; 38(5): 540-548. doi:10.1016/j.ctrv.2011.11.010.
- Ruffini E, et al: Recurrence of thymoma: analysis of clinicopathologic features, treatment, and outcome. J Thorac Cardiovasc Surg 1997; 113(1): 55-63. www.ncbi.nlm.nih.gov/pubmed/9011702. Accessed December 2, 2014.
- Chen G, et al: New WHO histologic classification predicts prognosis of thymic epithelial tumors: a clinicopathologic study of 200 thymoma cases from China. Cancer 2002; 95(2): 420-429. doi:10.1002/cncr.10665.
- Ruffini E, et al: Management of thymic tumors: a survey of current practice among members of the European Society of Thoracic Surgeons. J Thorac Oncol 2011; 6(3): 614-623. doi:10.1097/JTO.0b013e318207cd74.
- Korst RJ, et al: Adjuvant radiotherapy for thymic epithelial tumors: a systematic review and meta-analysis. Ann Thorac Surg 2009; 87(5): 1641-1647. doi:10.1016/j.athoracsur.2008.11.022.
- Forquer JA, et al: Postoperative radiotherapy after surgical resection of thymoma: differing roles in localized and regional disease. Int J Radiat Oncol Biol Phys 2010; 76(2): 440-445. doi:10.1016/j.ijrobp.2009.02.016.
- Loehrer PJ, et al: Cisplatin, doxorubicin, and cyclophosphamide plus thoracic radiation therapy for limited-stage unresectable thymoma: an intergroup trial. J Clin Oncol 1997; 15(9): 3093-3099. www.ncbi.nlm.nih.gov/pubmed/9294472. Accessed December 2, 2014.
- Girard N, Mornex F: The role of radiotherapy in the management of thymic tumors. Thorac Surg Clin 2011; 21(1): 99-105, vii. doi:10.1016/j.thorsurg.2010.08.011.
- Girard N, et al: Chemotherapy definitions and policies for thymic malignancies. J Thorac Oncol 2011; 6(7 Suppl 3): S1749–1755. doi:10.1097/JTO.0b013e31821ea5f7.
- Girard N: Thymoma: from chemotherapy to targeted therapy. Am Soc Clin Oncol Educ Book 2012: 475-479. doi:10.14694/EdBook_AM.2012.32.475.
- Berardi R, et al: Thymic neoplasms: an update on the use of chemotherapy and new targeted therapies. A literature review. Cancer Treat Rev 2014; 40(4): 495-506. doi:10.1016/j.ctrv.2013.11.003.
InFo ONCOLOGY & HEMATOLOGY 2015; 3(2): 16-20.









