For the prognosis of this autosomal recessive inherited disease, a diagnosis made in the neonatal period and the subsequent start of treatment are crucial – particularly with regard to the occurrence of complications such as hepatocellular carcinomas and neurocognitive impairments. Nitisinone is currently available as a pharmacological therapy, and certain dietary measures are also recommended.
In hepatorenal tyrosinemia type 1 (HT1), the formation of the enzyme fumarylacetoacetase (FAH), the last enzyme in tyrosine degradation, is impaired from birth. This is due to a biallelic pathogenic variant in the FAH gene. Due to the enzyme deficiency, the toxic metabolites fumarylacetoacetate, maleylacetoacetate and succinylacetone accumulate in the organism and damage the liver, kidneys and peripheral nervous system. As a result, untreated liver failure often occurs in the first year of life or, in a slower, chronic course, liver cirrhosis or hepatocellular carcinoma (HCC) develop [1]. Nitisinone is an active substance that intervenes in the early degradation cascade of tyrosine by competitively inhibiting the enzyme 4-hydroxyphenylpyruvate dioxygenase. This prevents the formation of toxic intermediate products. Treatment of all genotypes of the disease should be initiated as early as possible in order to prolong overall survival and prevent life-threatening organ manifestations. In Switzerland, nitisinone (Nityr®) was approved in the form of tablets in 2022 [2]. Starting treatment in the first few weeks of life not only prevents HCC in most cases, but also prevents liver and kidney dysfunction [3–5].
Before the era of nitisinone, the natural history of HT1 was usually fatal, 90% of HT1 patients died in the first two years of life and the only option was liver transplantation [1]. The availability of nitisinone (NTBC) has thus fundamentally changed the clinical course and outcome of people affected by HT1, according to the authors of the S2k guideline on the diagnosis and treatment of hepatorenal tyrosinemia (tyrosinemia type 1), which was updated in 2022 [1].
Newborn screening recommended
The decisive factor for the long-term prognosis, in particular the prevention of HCC, is to start treatment in the neonatal period. As the first symptoms do not appear in most patients until they are several months old, early diagnosis is not possible on purely clinical grounds [3]. Therefore, in case of clinical suspicion, the succinylazetone (SA) should be quantitatively determined from dried blood, serum/plasma and/or urine (“selective screening”). Determination of the tyrosine concentration alone has insufficient sensitivity and specificity and is therefore not recommended [1]. An SA measurement value above a defined cut-off is considered a positive screening result. A molecular genetic analysis of the FAH gene can be carried out to confirm the diagnosis (box). If the FAH geneis normal, the guideline recommends testing the GSTZ1 gene [1].
| Molecular genetic analyses The identification of biallelic pathogenic variants in the fumarylacetoacetase (FAH) geneconfirms the diagnosis of tyrosinemia type 1 [1]. DNA from blood cells or other body tissues (cheek swab) can be used for this analysis. RNA analysis from blood cells, liver biopsy samples or cultured fibroblasts is also possible. Single-gene analysis of the 14 coding exons of the FAH genein patients with a clear clinical/biochemical picture is a long-established method that detects pathogenic variants with an estimated sensitivity of >95% [1]. Nowadays, however, NGS(Next Generation Sequencing) techniques are mostly used as part of panel diagnostics or as part of WES (Whole Exome Sequencing) for FAH gene analysis– both for faster and more cost-effective diagnostics [16]. |
Start nitisinone therapy in the first weeks of life
The effects of nitisinone are based on the inhibition of the enzyme 4-hydroxyphenylpyruvate dioxygenase, which is involved in normal tyrosine degradation (Fig. 1). This prevents the formation of toxic metabolites. Side effects are rare, the most frequently reported being blood count disorders, eye disorders and an increased tyrosine concentration. Abruptly pausing or discontinuing nitisinone therapy can lead to “porphyria crises” and should be avoided [1, 6-8]. The therapeutic target value for nitisinone has not yet been well defined; the specialist literature states 20-60 μM as the therapeutic target value for nitisinone concentrations in plasma [3,9–11]. According to the authors of the guidelines, significantly lower concentrations are also possible without impairing metabolic control as measured by suppression of SA production [1]. The therapeutic target range for the tyrosine concentration in the blood is given as 200-800 μM, without there being any controlled, randomized, comparative studies on this [3,9–12].
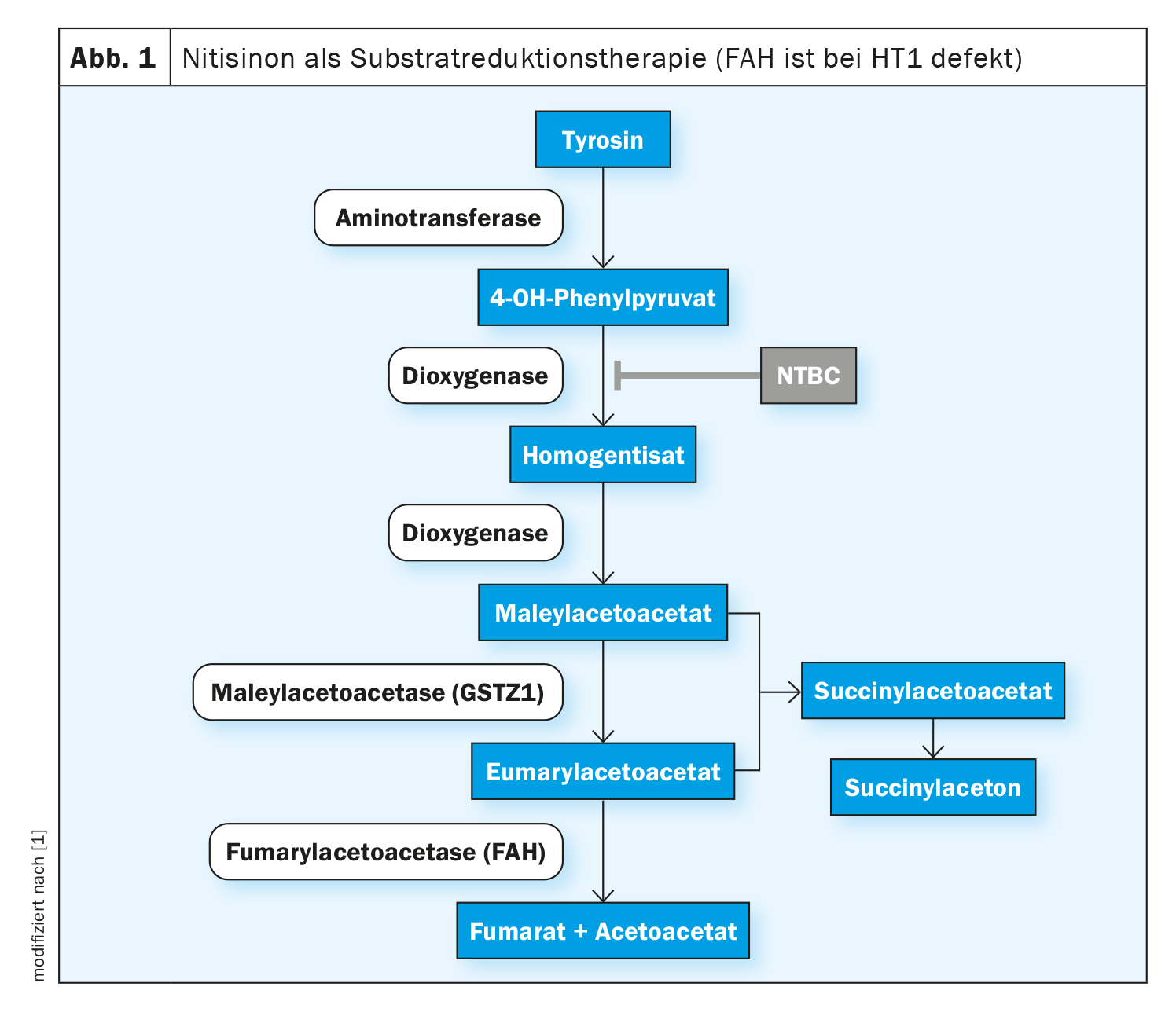
If possible, nitisinone therapy should be combined with a protein-reduced diet supplemented with a tyrosine- and phenylalanine-free amino acid mixture [13–15].
Diagnostic pitfalls – genetic analyses can help
The most important differential diagnosis of an elevated SA concentration is maleyl acetoacetate isomerase deficiency, a presumably benign metabolic abnormality that is not associated with liver dysfunction [1]. The guideline also points out that slightly elevated SA concentrations – depending on the cut-off value used – can also be temporary or due to mild forms of the disease that do not require treatment [1]. A positive screening result should be confirmed by one or more alternative analysis methods in addition to the control in the same sample material (dried blood). These include quantitative gas chromatography/mass spectrometry (GC/MS) analysis of organic acids in urine and the determination of SA in blood. If there is a strong suspicion, it is also advisable to check the liver function parameters. Abnormal findings in confirmation diagnostics can be further clarified by molecular genetic testing of the FAH gene. If this is inconspicuous, an analysis of the GSTZ1 gene (glutathione S-transferase zeta 1-1) can be performed, the deficiency of which is the cause of maleyl acetoacetate isomerase deficiency [1].
Literature:
- “S2k guideline: Diagnosis and treatment of hepatorenal tyrosinemia (tyrosinemia type 1)”, AWMF register number: 027-003, as of 09.06.2022.
- Swissmedic: Medicinal product information, www.swissmedicinfo.ch,(last accessed 06.02.2024)
- Mayorandan S, et al: Cross-sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J Rare Dis 2014; 9: 107.
- McKiernan PJ, Preece MA, Chakrapani A: Outcome of children with hereditary tyrosinaemia following newborn screening. Arch Dis Child 2015; 100(8): 738-741.
- Bartlett DC, et al: Early nitisinone treatment reduces the need for liver transplantation in children with tyrosinaemia type 1 and improves post-transplant renal function. J Inherit Metab Dis 2014; 37(5): 745-752.
- Önenli Mungan N, et al: Tyrosinemia type 1 and irreversible neurologic crisis after one month discontinuation of nitisone. Metab Brain Dis 2016; 31(5): 1181-1183.
- Schlump JU, et al: Severe neurological crisis in a patient with hereditary tyrosinaemia type I after interruption of NTBC treatment. J Inherit Metab Dis 2008; 31 Suppl 2: S223-S225.
- Uçar HK, et al: A Case Report of a Very Rare Association of Tyrosinemia type I and Pancreatitis Mimicking Neurologic Crisis of Tyrosinemia Type I. Balkan Med J 2016; 33(3): 370-372.
- Chinsky JM, et al: Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med 2017; 19(12): doi:10.1038/gim.2017.101
- de Laet C, et al: Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis 2013; 8: 8. Published 2013 Jan 11. doi:10.1186/1750-1172-8-8
- Alvarez F, Mitchell GA: Tyrosinemia and Liver Transplantation: Experience at CHU Sainte-Justine. Adv Exp Med Biol 2017; 959: 67-73.
- De Laet C, et al: Neuropsychological outcome of NTBC-treated patients with tyrosinaemia type 1. Dev Med Child Neurol 2011; 53(10): 962-964.
- Morrow G, Angileri F, Tanguay RM: Molecular Aspects of the FAH Mutations Involved in HT1 Disease. Adv Exp Med Biol 2017; 959: 25-48.
- Morrow G, Tanguay RM: Biochemical and Clinical Aspects of Hereditary Tyrosinemia Type 1 Adv Exp Med Biol 2017; 959: 9-21.
- van Spronsen FJ, et al: Dietary Considerations in Tyrosinemia Type I. Adv Exp Med Biol 2017; 959: 197-204.
- Blackburn PR, et al: Silent Tyrosinemia Type I Without Elevated Tyrosine or Succinylacetone Associated with Liver Cirrhosis and Hepatocellular Carcinoma. Hum Mutat 2016; 37(10): 1097-1105.
FAMILY PHYSICIAN PRACTICE 2024; 19(2): 36-37