
The severity and prevalence of iron deficiency in chronic renal failure parallel its stage and are multifactorial in origin. In patients with renal insufficiency, iron homeostasis is substantially altered, so that optimal iron parameters are defined differently than in renal healthy subjects. This has implications for aspects of medical history, examinations to be performed, and limits and targets. This article addresses this issue and analyzes iron supplementation in oral and parenteral forms.
The element iron is an essential and quantitatively the most important trace element for almost all living beings. Occurring in animals and humans in divalent and trivalent forms, iron is mainly involved in oxygen binding and electron transfer. These physicochemical properties result in its main functions as oxygen binder and transporter, catalyst of redox reactions in the cellular respiratory chain, and active component of numerous non-heme enzymes. However, the ability of free iron ions to form highly reactive oxygen species (e.g. hydroxyl radicals) from hydrogen peroxide (so-called Fenton reaction) and to cause lipid peroxidation harbors considerable cytotoxic potential. For this reason, the majority of iron, both intracellular and extracellular, is present in redox-inactive forms bound to specialized proteins [1].
Iron homeostasis
The adult human body contains about 3.5-5 g of elemental iron [2]. This is present almost exclusively in bound form as storage and functional iron. The storage pool (0.8-1.2 g) consists of the protein complex ferritin, an approximately 450 kDa protein filled with iron(III) hydroxide oxide, and the strictly intracellular hemosiderin, which is a slow and difficult-to-mobilize storage form of denatured ferritin. The liver, spleen and bone marrow are particularly rich in storage iron. Although the proportion of extracellular ferritin is comparatively minor, its serum concentration appears to correlate well with the intracellular iron reserve in normal cases.
Functional iron includes hemoglobin (2.5-3 g), which is the most important in terms of quantity, myoglobin (150 mg), intracellular iron (80 mg), and iron-containing enzymes (10 mg). Transport iron bound to transferrin, a glycoprotein about 80 kDa in size, and circulating (4 mg) forms relatively only a small fraction of the total amount. Thus, the rapidly exchangeable iron available for the re-synthesis of heme and iron-containing enzymes (25 mg/day) is recycled several times per day. This occurs through the breakdown of heme groups in the reticuloendothelial system. In this context, transferrin saturation, the ratio of serum iron to total iron binding capacity (transferrin), represents an indirect measure of available iron [1, 3].
Figure 1 provides an overview of the amount of iron present and its distribution in healthy adults.
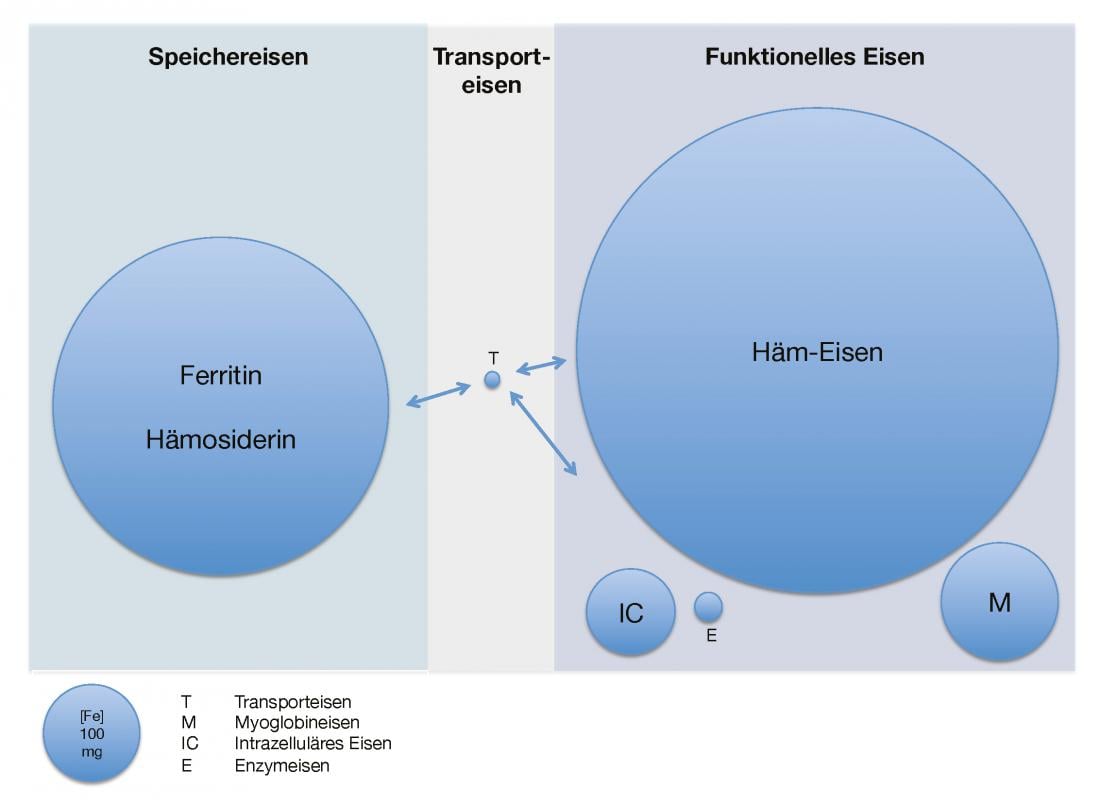
Fig. 1: Available iron quantity and distribution in healthy adults
Iron distribution in the body under quantitative aspects. T stands for transferrin-bound iron, M for myoglobin iron, IC for intracellular iron, and E for iron contained in enzymes.
Due to its dual property as an indispensable trace element and a highly potent organ and cell toxin, the balance of iron and its compartmentalization in the body is strictly regulated. Unlike polyvalent cations such as calcium, iron homeostasis is regulated exclusively by absorption, since normal losses cannot be controlled and no additional increase in iron excretion is possible. Thus, in healthy individuals in steady state, daily enteral iron intake (1-2 mg in men, 2-3 mg in menstruating women) covers natural losses through exfoliation of skin and mucosal cells, menstruation in women of childbearing age, and trace amounts through stool and urine.
In mammals, the poorly absorbable iron(III) is reduced to the more soluble iron(II) by duodenal cytochrome B at the apical pole of enterocytes. Then, intracellular uptake into the enterocyte occurs through the divalent metal transporter (DMT1). Iron can also be taken up by a parallel and very efficient mechanism as hemin, a degradation product of heme and myoglobin abundant in meat products, through the heme carrier protein 1 (HCP1) transporter. Iron(II) is now exported by the basolateral ferroportin and immediately oxidized by ceruloplasmin to iron(III), which binds to transferrin. However, the rate of absorption of orally ingested iron is low, averaging 6% in males and about twice that in young females. An increase when the system is maximally stimulated (iron deficiency) can only be as high as 20%. A key role in this fine regulation is played by the peptide hormone hepcidin, discovered about 13 years ago. This binds and induces degradation of the iron exporter ferroportin in duodenal enterocytes, reticuloendothelial macrophages and hepatocytes and prevents iron from being delivered to transferrin. This results in low enteric iron absorption and decreased iron delivery from ferritin to transferrin, termed reticuloendothelial blockade. Circulating iron and inflammatory mediators stimulate hepcidin production, whereas hypoxemia and ESA administration have an inhibitory effect [1, 3-5].
Iron deficiency in chronic renal failure
One of the most important features of chronic renal failure is renal anemia, which is associated with decreased quality of life, increased cardiovascular morbidity, and all-cause mortality [6, 7]. In addition to relative erythropoietin deficiency [8, 9], uremic-related erythropoietin resistance [10], and markedly shortened erythrocyte survival in the uremic milieu [11], absolute and functional iron deficiency should be mentioned as an important causative factor. Both are also important factors of erythropoietin resistance, which is characterized by the high need for erythropoiesis-stimulating agents (ESAs) and difficulty or even impossibility to reach the therapeutic target without the need for transfusion [12, 13].
The prevalence and severity of iron deficiency in chronic renal failure parallel its stage and are multifactorial in origin. Inappetence frequently observed in this population, prescribed dietary phosphate restriction, and high and generalized use of proton pump inhibitors and calcium-containing phosphate binders lead to decreased iron intake and absorption [14, 15]. Due to uremic thrombopathy and gastritis, there are additional gatrointestinal losses. In parallel, with increasing renal insufficiency, there is increased formation and accumulation of hepcidin due to the uremia-associated proinflammatory state and decreased clearance of peptide hormones. This particular constellation explains the frequently observed functional iron deficiency of various degrees, characterized by high ferritin levels and low to normal transferrin saturation levels [4, 12, 15, 16]. Accordingly, in renal insufficient patients, the ferritin value for defining iron deficiency is set higher than in renal healthy patients at 100-200 ng/ml [6, 7, 15, 17]. An alternative formulation for this would be that optimal iron utilization in these patients occurs at the expense of increased storage iron. Thus, serum ferritin levels below 100 ng/ml are found to have a suboptimal response to ESA and insufficient stores [18].
In contrast, almost all renal failure patients, including hemodialysis patients, have sufficient intramedullary iron reserves when serum ferritin levels are above 300 ng/ml [19]. However, Mirahmadi et al. show bioptically decades ago that at serum ferritin levels above 124 ng/ml certain hemodialysis patients already show signs of iron overload [20]. The whole situation becomes even more complex when considering the fact that certain hemodialysis patients with high serum ferritin levels (up to more than 1200 ng/ml) continue to show a positive response to parenteral iron administration, especially when transferrin saturation is suboptimal at less than 20% [21, 22].
These figures clearly demonstrate that the general assessability of these tests should be put into perspective [23]. In the case of rapid mobilization of the iron reserve, as is the case, for example, in the context of newly started or intensified ESA therapy, iron replenishment from the reserves may be insufficient, resulting in so-called iron-limited erythropoiesis. This can occur independently of functional iron deficiency and is characterized by decreased hemoglobin content of erythrocytes and their precursors. The percentage of hypochromic erythrocytes (% HRC) and reticulocyte hemoglobin content (CHr) are well-established indicators of functional iron deficiency and iron-limited eythropoiesis. Thus, a CHr below 29 pg/cell in patients on ESA therapy is indicative of functional iron deficiency. Reticulocyte hemoglobin equivalent (Ret-HE) below 30.6 pg/cell, a counterpart of CHr measured by an alternative method, is itself highly predictive of a good response to intravenous iron administration in hemodialysis patients treated with ESA. Newer and more precise parameters of functional iron deficiency and iron-limited erythropoiesis, such as erythrocytic zinc protoporphyrin (ZPP) and soluble transferrin receptor (sTFR), are currently not recommended as standard tests due to limited availability, higher costs, and still insufficient experience in renal insufficient patients [22].
According to the above criteria, the prevalence of iron deficiency in non-dialysis-dependent renal failure patients (CKD stages 3 and 4) is more than 50% [15]. The situation in hemodialysis patients is even more critical. Apart from factors already mentioned, there is a considerable loss of iron of 1-3 g (at normal hematocrit, 1 ml of blood contains about 0.5 mg of iron) due to numerous blood withdrawals and regular blood sequestration in the filter and dialysis equipment. This virtually leads to iron deficiency in all patients if the losses are not replaced by adequate substitution [10].
Isolated iron deficiency without anemia (EoA), an otherwise increasingly well-defined entity in renally competent patients [24, 25], is rare in renally insufficient patients. Often, these are iatrogenic situations in which patients do not receive iron supplementation or even phlebotomy for a prolonged period of time due to spontaneously high hematocrit levels. Inadequate therapy of renal anemia with high-dose ESA and lack of or inadequate iron substitution may lead to the same picture. Typical general symptoms of EoA such as fatigue, attention deficits, impaired thermoregulation, and hair loss are absent or masked by the foreground uremic symptoms. Only restless legs syndrome (RLS) appears to be associated with functional iron deficiency in this population, independent of anemia level [26].
Iron supplementation and monitoring in renal insufficiency.
All renal patients should be evaluated for the presence of anemia. The prevalence of renal anemia increases markedly from CKD stage 3 and requires appropriate workup [6, 7, 27]. In addition to a targeted history and examination, this includes at least a complete blood count with erythrocyte indices and absolute reticulocyte count and a serum vitamin B12 and folate level. Iron status should be assessed for storage iron (serum ferritin) and rapidly mobilizable iron (transferrin saturation and/or a reticulocyte hemoglobin content) [6, 7].
Iron substitution is recommended in principle in all renal failure dialyzed and non-dialyzed patients with anemia and ESA therapy. In addition, parenteral iron administration can avoid or at least delay the need for ESA therapy in nondialyzed patients and contributes to a substantial reduction in ESA requirements in hemodialysis patients, which is associated with financial and possibly health benefits [6, 7, 13, 28].
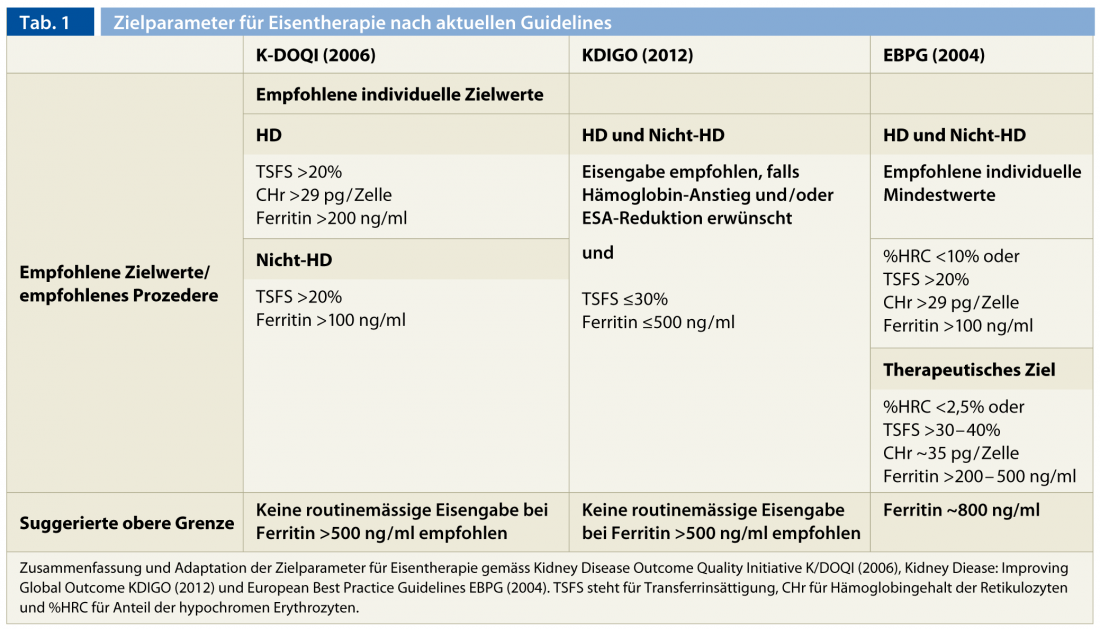
Although not uniformly defined by expert groups, a consensus on target values can be extracted. A minimum value of 200 ng/ml for serum ferritin and 20% for transferrin saturation are unanimously accepted. Alternatively, target values for CHr (>29 pg/cell) and % HCR (<10%) are defined in addition to transferrin saturation. These minimum requirements do not per se represent population-level target values, which are naturally set higher here.
It remains more complicated to make a clear statement about the upper limits with regard to the immediate and the still poorly studied late complications of parenteral iron therapy [29–31]. Signs of liver toxicity and increased mortality and susceptibility to infection have been observed in hemodialysis patients with iron overload [15, 29, 32]. Generally, iron administration is not recommended for ferritin levels above 500 ng/ml [6, 7, 17]. Furthermore, parenteral iron administration should be avoided in active infections and fever due to the deleterious effects on the immune system and decreased transport capacity with inflammation [7]. Persistent functional iron deficit requires additional workup of cryptogenic infection or neoplasia. If both are excluded, optimization of hemodialysis treatment and controlled parenteral iron administration can be used to try to overcome this condition. In the near future, it is hoped that the therapeutic use of hepcidin inhibitors will enable “uremic iron blockade” to be treated even more efficiently. Table 1 summarizes target parameters for iron therapy according to currently available guidelines.
The therapeutic effect should be on a regular basis to ensure continuous optimal iron reserves and to avoid toxic side effects due to uncontrolled accumulation of storage iron (from about 500-800 ng/ml ferritin) and/or exceeding the serum transport capacity with deleterious increase of free iron in serum (TSFS >60%) [33]. It is important to note that the assessability of iron parameters is severely impaired immediately after parenteral iron administration and an interval of at least one week between administration and measurement should be maintained. Monthly controls during the saturation phases or at the beginning of ESA therapy are recommended. In stable cases and in patients without ESA therapy, three-monthly checks are sufficient.
Oral iron substitution
Despite low price and ease of use, oral iron substitution has several disadvantages. Iron taken perorally can lead to oxidative damage in the gastrointestinal tract, which can manifest clinically with vomiting, dyspepsia, and diarrhea. The resulting poor compliance can be substantial, as high as 70% in certain collectives [34]. Furthermore, the efficacy of oral iron supplementation remains very low for the reasons already mentioned.
More effective replenishment of iron reserves and better response to ESA can only be achieved with the parenteral form [14]. Even in the case of previously normalized iron reserves, peroral administration was not able to maintain sufficient iron reserves in the longer term [35]. When using high-dose iron salt-containing preparations (iron[II] anion), the passive and uncontrolled diffusion of iron can lead to a significant increase in free iron, so-called non-transferrin bound iron (NTBI), due to exhaustion of local transport mechanisms, which can also result in systemic toxicity. In general, oral substitution should be limited to nondiaylse patients without ESA therapy and without severe iron deficiency under regular evaluation of efficacy and tolerance [6, 7].
Parenteral iron substitution
The high iron demand, especially in hemodialysis patients, exceeds the capacity of enteral iron uptake and delivery from the storage pool, which is already impaired in the context of renal insufficiency, and explains the clear therapeutic superiority of parenteral administration over peroral administration. For this reason, it is recommended as the standard form of administration in all renal failure patients. Three parenteral iron preparations are currently available in Switzerland (Table 2).
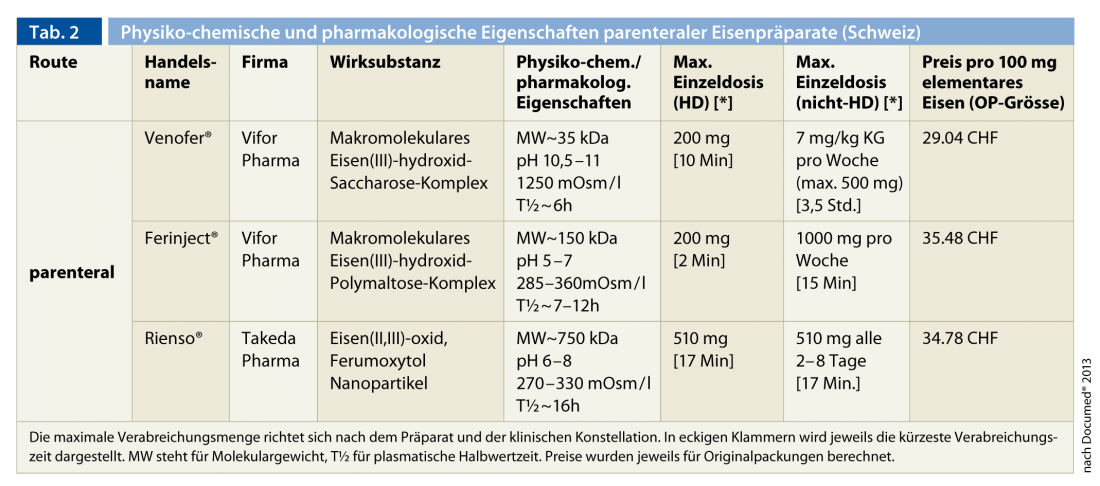
In chronological order of approval, these are Venofer® (macromolecular iron(III) hydroxide sucrose complex), Ferinject® (macromolecular iron(III) hydroxide polymaltose complex) and Rienso® (iron(II, III) oxide (ferumoxytol) nanoparticles). The low- and high-molecular-weight preparations containing dextran are no longer on the market due to the potentially life-threatening undesirable side effects (anaphylaxis). The experience gained over decades with the former preparation (Venofer®) is considerable. The two newer preparations allow the administration of higher single doses due to their physicochemical properties (pH and osmolality) and especially due to the higher stability of the iron complex. This is a significant advantage for outpatients not on hemodialysis. In all cases, the incidence of serious life-threatening side effects is very low. Cross-reactions with the preparations containing dextran, which are not available in Switzerland, are not to be expected [15, 36]. Sporadic hypotensive episodes, bronchospasm, skin reaction, myalgias and arthralgias or fever are observed, especially with rapid administration.
The therapy is divided into a replenishment phase and a maintenance phase. Initial iron requirements can be calculated using the Ganzoni formula:
Total iron deficit (mg) = [Soll-Hb – Ist-Hb (g/dl)] × body weight (kg) × 2.4 + reserve iron (mg).
This amount can then be delivered fractionally (Venofer® and Ferinject®) or as a single dose (Ferinject® and Rienso®), depending on the preparation, the severity of the renal insufficiency and the form of administration. The maintenance dose is based on the therapeutic effect achieved and the regularly monitored iron parameters.
CONCLUSION FOR PRACTICE
- Absolute and functional iron deficiency is common in chronic renal failure and is significantly involved in the development of renal anemia.
- Iron supplementation is a key component in the treatment of renal anemia. Optimization of iron parameters allows therapeutic target values to be achieved in most patients and minimizes the consumption of ESA, which is associated with cost reduction and potentially better outcome.
- Iron homeostasis is substantially altered in renal failure, so optimal iron parameters are defined differently than in renal health.
- Nowadays, parenteral administration is superior to peroral administration due to better efficacy and tolerability. More modern parenteral iron preparations are safe and greatly facilitate iron substitution.
- Because of the lack of long-term outcome studies and existing evidence for deleterious effects of iron overdose in this population, systematic iron administration should be performed only while adhering to target levels.
Robert M. Kalicki, M.D.
Literature:
- Dunn LL, Suryo Rahmanto Y, Richardson DR: Trends Cell Biol 2007; 17, 93.
- Löffler G: Biochemistry and Pathobiochemistry. 7 edition. Springer-Verlag: Berlin 2002.
- Andrews NC: Blood 2008; 112: 219.
- Babitt JL, Lin HY: Am J Kidney Dis 2010; 55: 726.
- Nemeth E, et al: Science 2004; 306: 2090.
- Am J Kidney Dis 2006; 47: S16.
- Kidney International Supplements 2012; 2: 279.
- Jacobson LO, Goldwasser E, Fried W, Plzak L: Nature 1957; 179: 633.
- McGonigle RJ, Wallin JD, Shadduck RK, Fisher JW: Kidney Int 1984; 25: 437.
- Besarab AF, in: Anemia in renal disease. Lippincott Williams and Wilkins: Philadelphia 2007; 2406-2430.
- Eschbach JW: Kidney Int 1989; 35: 134.
- Babitt JL, Lin HY: J Am Soc Nephrol 2012; 23: 1631.
- Coyne DW, Auerbach M: Am J Hematol 2012; 85: 311.
- Fudin R, Jaichenko J, Shostak A, Bennett M, Gotloib L: Nephron 1998; 79: 299.
- Macdougall IC, Geisser P: Iran J Kidney Dis 2012; 7: 9.
- Ashby DR, et al: Kidney Int 2009; 75: 976.
- Nephrol Dial Transplant 2004; 19 [Suppl 2]: ii6.
- Fernandez-Rodriguez AM, et al: Am J Kidney Dis 1999; 34: 508.
- Aljama P, et al: Clin Nephrol 1978; 10: 101.
- Mirahmadi KS, et al: JAMA 1977; 238: 601.
- Chang CH, Chang CC, Chiang SS: Clin Nephrol 2002; 57: 136.
- Thomas DW, et al: Br J Haematol 2013.
- Kalantar-Zadeh K, et al: Am J Kidney Dis 1995; 26: 292.
- Bruner AB, Joffe A, Duggan AK, et al: Lancet 1996; 348: 992.
- Rowland TW, Deisroth MB, Green GM, et al: Am J Dis Child 1988; 142: 165.
- Sloand JA, Shelly MA, Feigin A, et al: Am J Kidney Dis 2004; 43: 663.
- Bethesda MD, The National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases 2004.
- Solomon SD, et al: N Engl J Med 2010; 363: 1146.
- Kalantar-Zadeh K, et al: J Am Soc Nephrol 2005; 16: 3070.
- Feldman HI, et al: J Am Soc Nephrol 2002; 13: 734.
- Feldman HI, et al: J Am Soc Nephrol 2004; 15: 1623.
- Vaziri ND: Am J Kidney Dis 2013.
- Hershko C, Graham G, Bates GW, et al: Br J Haematol 1978; 40: 255.
- Kruske SG, Ruben AR, Brewster DR: J Paediatr Child Health 1999; 35: 153.
- Macdougall IC, et al: Kidney Int 1996; 50: 1694.
- Auerbach M, Ballard H: Hematology Am Soc Hematol Educ Program 2010; 338.









