Existen diversas mutaciones que pueden subyacer a la distrofia hereditaria de retina. Gracias al desarrollo de los modernos métodos de pruebas genéticas moleculares, en la actualidad es posible identificar la mayoría de las causas genéticas. Existe una innovadora terapia génica para pacientes con distrofia de retina debida a mutaciones bialélicas del RPE65.
El término paraguas “distrofias hereditarias de la retina” (IRD; enfermedades hereditarias de la retina) engloba un grupo heterogéneo de enfermedades con fenotipos superpuestos que se caracterizan por la degeneración progresiva y la disfunción de la retina [1]. Lo que estas enfermedades tienen en común es que afectan a toda la retina durante el curso de la enfermedad y que la visión se deteriora lenta y progresivamente a lo largo de la vida de una persona. La IRD puede estar causada por mutaciones en >250 genes diferentes [2]. La patogénesis molecular, así como el cuadro clínico y el curso de la enfermedad, están definidos de forma significativa por el tipo y la naturaleza del defecto genético [3]. Dependiendo del gen mutado, pueden producirse diversos trastornos funcionales y del desarrollo de la retina, como alteraciones del metabolismo retiniano, de las estructuras celulares o de la fototransducción. La forma más común de IRD es la retinosis pigmentaria, pero también existen la amaurosis congénita de Leber y la distrofia retiniana de aparición temprana (EORD) [2].
El diagnóstico precoz es crucial
En la retinosis pigmentaria, la degeneración de los bastones se produce en las primeras fases de la enfermedad, lo que provoca ceguera nocturna. Los síntomas característicos de la amaurosis congénita de Leber incluyen una reducción drástica de la agudeza visual y defectos del campo visual. El diagnóstico correcto y precoz de la enfermedad es muy importante, así como una descripción morfológica y funcional detallada del estado actual de la retina en función de las mutaciones subyacentes en el gen RPE65. Esto se debe a que la rápida y grave progresión de la enfermedad hace necesaria una intervención precoz mientras las células del epitelio pigmentario de la retina como células diana y también los fotorreceptores sigan presentes. En el marco del diagnóstico funcional oftalmológico y, especialmente, del diagnóstico por imagen no invasivo (por ejemplo, tomografía de coherencia óptica, fondo de ojo o autofluorescencia en el infrarrojo cercano), es posible la descripción clínica del fenotipo [5,6]. Para identificar la causa genética de las distrofias hereditarias de retina, es indispensable utilizar métodos de genética molecular como la secuenciación de nueva generación.
Las imágenes de autofluorescencia proporcionan hallazgos importantes
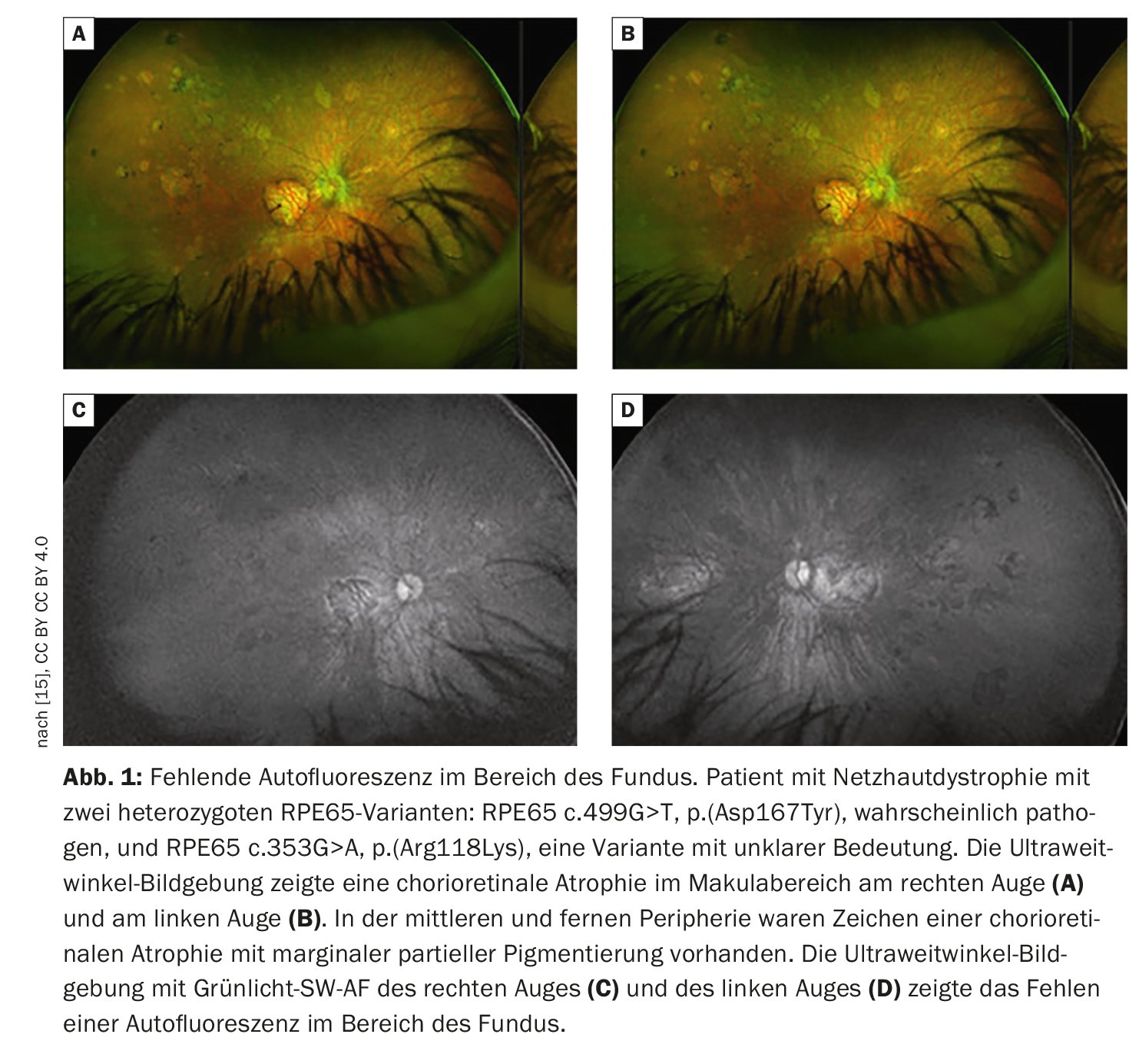
La imagen retiniana proporciona una visión más detallada de la patología retiniana que el examen funduscópico. Además de la fotografía convencional del fondo de ojo en color, la tomografía de coherencia óptica (OCT) de alta resolución y la autofluorescencia del fondo de ojo, que permite visualizar los fluoróforos del fondo ocular mediante luz de onda corta, son los métodos más consolidados. La autofluorescencia del fondo del ojo muestra la distribución de los fluoróforos del fondo del ojo, utilizando principalmente luz de excitación de onda corta en la gama azul o verde. En los pacientes con mutaciones en el gen RPE65, la ausencia o al menos la reducción de la autofluorescencia en la región del fondo de ojo (Fig. 1) es una característica clínicamente significativa debida al metabolismo defectuoso de los retinoides en los fotorreceptores y las células del EPR [9]. Por ejemplo, en la retinosis pigmentaria suele haber un anillo concéntrico de autofluorescencia aumentada para el que no existe un correlato visible funduscópicamente [10–12]. Aunque no se conoce con exactitud el origen de este fenómeno, los estudios con OCT han demostrado que el anillo corresponde a la pérdida de la banda elipsoidal y a un grave adelgazamiento o incluso pérdida de la capa fotorreceptora [10].
Pruebas genéticas moleculares para la detección de la causa genética
Los fenotipos de las variantes de distintos genes suelen solaparse y es posible una variabilidad considerable en las variantes de un mismo gen. Por lo tanto, las pruebas genéticas moleculares son esenciales y una base importante para la selección de la opción terapéutica adecuada [6]. En el 55-80% de los casos, la causa genética de las distrofias hereditarias de retina puede identificarse mediante pruebas genéticas moleculares [4]. En los pacientes con mutaciones bialélicas en el gen RPE65, la isomerasa RPE65 no se expresa en absoluto o se expresa como una proteína no funcional en el epitelio pigmentario de la retina (EPR). Por definición, para el diagnóstico de la distrofia de retina asociada a la mutación RPE65 bialélica, ambas copias del gen RPE65 deben estar presentes como una o dos variantes patogénicas distintas [8]. El fenotipo clínico en el que se manifiestan las mutaciones bialélicas del RPE65 suele estar asociado a la amaurosis congénita de Leber o a la retinosis pigmentaria. La falta de RPE65 isomerasa funcional significa que el todo-trans-retinal producido en la cascada de fototransducción no se convierte en 11-cis-retinal, lo que perjudica la regeneración del pigmento visual rodopsina [1]. Esta alteración del metabolismo retiniano hace que los fotorreceptores pierdan su capacidad de responder a los estímulos luminosos. Además, la acumulación de intermediarios tóxicos conduce a la muerte de las células epiteliales pigmentarias de la retina, lo que provoca una disfunción adicional de los fotorreceptores. Este efecto es más pronunciado en los bastones fotorreceptores que en los conos fotorreceptores, ya que estos últimos aún disponen de una vía metabólica alternativa para la regeneración del pigmento visual a través de las llamadas células de Müller [1].
Fármaco de terapia génica como opción de tratamiento para la mutación en el gen RPE65
La idea básica es que añadiendo una copia correcta mediante la terapia génica retiniana, se puede restaurar la función de la retina. Los vectores virales recombinantes adenoasociados (AAV) son muy adecuados para realizar la transferencia de genes, ya que permiten una expresión duradera y específica de cada tipo celular en la retina [9]. Voretigen Neparvovec es un vector de transferencia genética que utiliza la cápside de un vector viral adenoasociado de serotipo 2 (AAV2) como vehículo de transporte del ADNc de la proteína de 65 kDa específica del epitelio pigmentario de la retina humana (hRPE65) a la retina [13].
| Neparvovec preetigénico con mutación RPE65 bialélica La eficacia de Luxturna® (voretigen neparvovec) se estudió en un total de 41 pacientes, todos ellos con distrofia hereditaria de retina [13]. En el estudio principal participaron 11 adultos (36%) y 20 niños mayores de 4 años (64%). La edad media era de 15 años. Para probar la eficacia de Luxturna® en el estudio principal, se midió la visión funcional. Se compone de la agudeza visual, el campo visual y la percepción y/o visión con poca luz. Tras un año de tratamiento, los pacientes del brazo de tratamiento con Luxturna® pudieron completar un recorrido con mayor precisión, más rápidamente y en condiciones de menor iluminación. La mejora de la visión funcional se mantuvo durante un periodo de al menos tres años. Se está realizando un seguimiento de 15 años de los 41 participantes en el estudio. |
Luxturna® es un medicamento de terapia génica que contiene el principio activo voretigen neparvovec y está aprobado en Suiza desde febrero de 2020 para el tratamiento de adultos y niños con pérdida de visión debida a distrofia hereditaria de retina con mutación RPE65 bialélica (box) [13]. El tratamiento con Luxturna® requiere que la mutación RPE65 se haya confirmado mediante pruebas genéticas y que aún existan suficientes células funcionales en la retina del paciente [13]. Luxturna® se aplica microquirúrgicamente bajo la retina [13,14]. En Suiza, existe un centro exclusivo para esta terapia génica en el Hospital Universitario de Basilea (USB) [14]. El Prof. Dr. med. Hendrik Scholl es jefe y médico jefe de la Clínica Oftalmológica de la USB, el Prof. Dr. med. Christian Prünte es médico jefe clínico de la misma y se ha especializado, entre otras cosas, en microcirugía. En noviembre de 2022, la USB anunció que un equipo de doce miembros dirigido por los profesores Scholl y Prünte había realizado la primera operación de microcirugía en Suiza para la aplicación de Luxturna®. El paciente de 51 años con distrofia hereditaria de retina se sometió a un tratamiento en su segundo ojo en marzo de 2022, tras una intervención quirúrgica en el otro ojo un año antes [14]. Ambas intervenciones se desarrollaron de forma óptima.
Mensajes para llevarse a casa
- Hasta la fecha, se conocen más de 250 mutaciones que subyacen a la distrofia hereditaria de retina. En el 55-80% de los casos, la causa genética puede identificarse mediante pruebas genéticas moleculares [4].
- El defecto genético subyacente define la patogénesis molecular, el cuadro clínico y el curso de la enfermedad. Las variantes del gen RPE65 son la causa del 0,6-6% de todos los casos de retinosis pigmentaria y del 3-16% de los casos de amaurosis congénita de Leber/distrofia retiniana de aparición precoz [2].
- En la imagen retiniana, las mutaciones en el gen RPE65 se caracterizan por una autofluorescencia ausente o reducida en el fondo de ojo.
- Luxturna® (voretigene neparvovec) es la primera terapia génica disponible para el tratamiento de la distrofia retiniana hereditaria con mutación RPE65 bialélica [13].
Literatura:
- Dias MF, et al: Genética molecular y terapias emergentes para la retinosis pigmentaria: Investigación básica y perspectivas clínicas. Progress in Retinal and Eye Research 2017 (63): 107-131.
- Aoun M, et al: Distrofias hereditarias de retina debidas a variantes del RPE65: Del diagnóstico genético a la terapia. Compass Ophthalmol 2021; 7: 115-123.
- Nash BM, et al: Distrofias retinianas, aplicaciones genómicas en el diagnóstico y perspectivas terapéuticas. Pediatría traslacional 2015; 4 (2): 139-163.
- Kohl S, Biskup S: Pruebas de diagnóstico genético en las distrofias hereditarias de retina. Oftalmología Clin Monbl 2013; 230(3): 243-246.
- Bolz HJ: Análisis diagnósticos de genes de distrofia retiniana: estado actual y perspectivas. Oftalmología Clin Monbl 2021; 238: 261-266.
- Kellner U, et al.: Diagnóstico de las distrofias hereditarias de retina. Importancia del diagnóstico genético molecular desde la perspectiva del paciente. Oftalmología 2022; 119: 820-826.
- Dockery A, et al: Aplicaciones de la secuenciación de próxima generación para las enfermedades hereditarias de la retina. Int J Mol Sci 2021; 22: 5684.
- Blue Cross and Blue Shield de Kansas City (BCBSKC) 2018. Terapia génica para la distrofia hereditaria de retina: Norma número 2.04.144.
- Stieger K, Lorenz B: Terapia génica para enfermedades degenerativas de la retina, www.kaden-verlag.de/fileadmin/images/ZPA_direkt/CME_ZPA_12-09.pdf,(última consulta: 04.04.2023)
- Birtel J, et al: Diagnóstico de las enfermedades hereditarias de la retina Klin Monatsbl Augenheilkd 2021; 238: 249-260.
- Robson AG, et al: Serial imaging and structure-function correlates of high-density rings of fundus autofluorescence in retinitis pigmentosa. Retina 2011; 31: 1670-1679.
- Lima LH, et al: Constricción progresiva del anillo hiperautofluorescente en la retinosis pigmentaria. Am J Ophthalmol 2012; 153: 718-727.
- Información sobre medicamentos, www.swissmedicinfo.ch,(último acceso 04.04.2023)
- “Primer uso de la terapia génica contra la ceguera en Suiza”, www.unispital-basel.ch/newscenter/zuweisenden-blog/15-07-2022,(última consulta: 04.04.2023)
- Bjeloš M, et al.: RPE65 c.353G>A, p.(Arg118Lys): Una nueva mutación puntual asociada a la retinosis pigmentaria y la atrofia macular. Revista Internacional de Ciencias Moleculares. 2022; 23(18):10513. www.mdpi.com/1422-0067/23/18/10513,(última consulta: 04.04.2023).
PRÁCTICA GP 2023; 18(4): 46-47