In autosomal recessive diseases such as cystic fibrosis (CF), one is dealing with the inheritance of two mutated genes. Previously, CFTR* expression levels of 50% were thought to be sufficient for maintenance of health. However, some studies have suggested that even carriers of just one mutated gene are already at increased risk of developing manifestations of the disease.
* Cystic Fibrosis Transmembrane Conductance Regulator
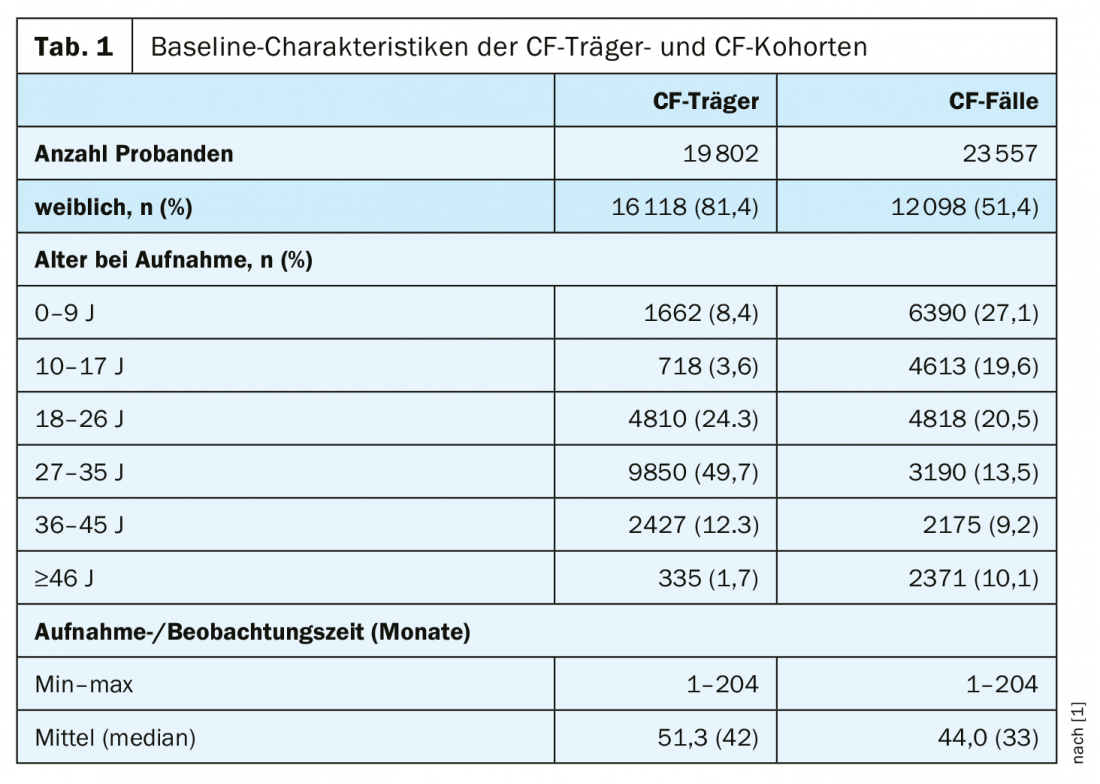
However, these studies focused on single aspects and included a relatively small number of subjects, no controls, and did not compare risk between CF carriers and CF patients. A team led by U.S. epidemiologist Dr. Aaron C. Miller of the College of Public Health at the University of Iowa has now conducted a large population-based retrospective cohort study that included 19,802 carriers of a mutated CFTR gene. Each carrier was compared with five matched controls (n=99,010). For the CF cohort, they identified 23,557 patients with CF who were matched to a non-CF cohort of 117,762 subjects (Table 1).
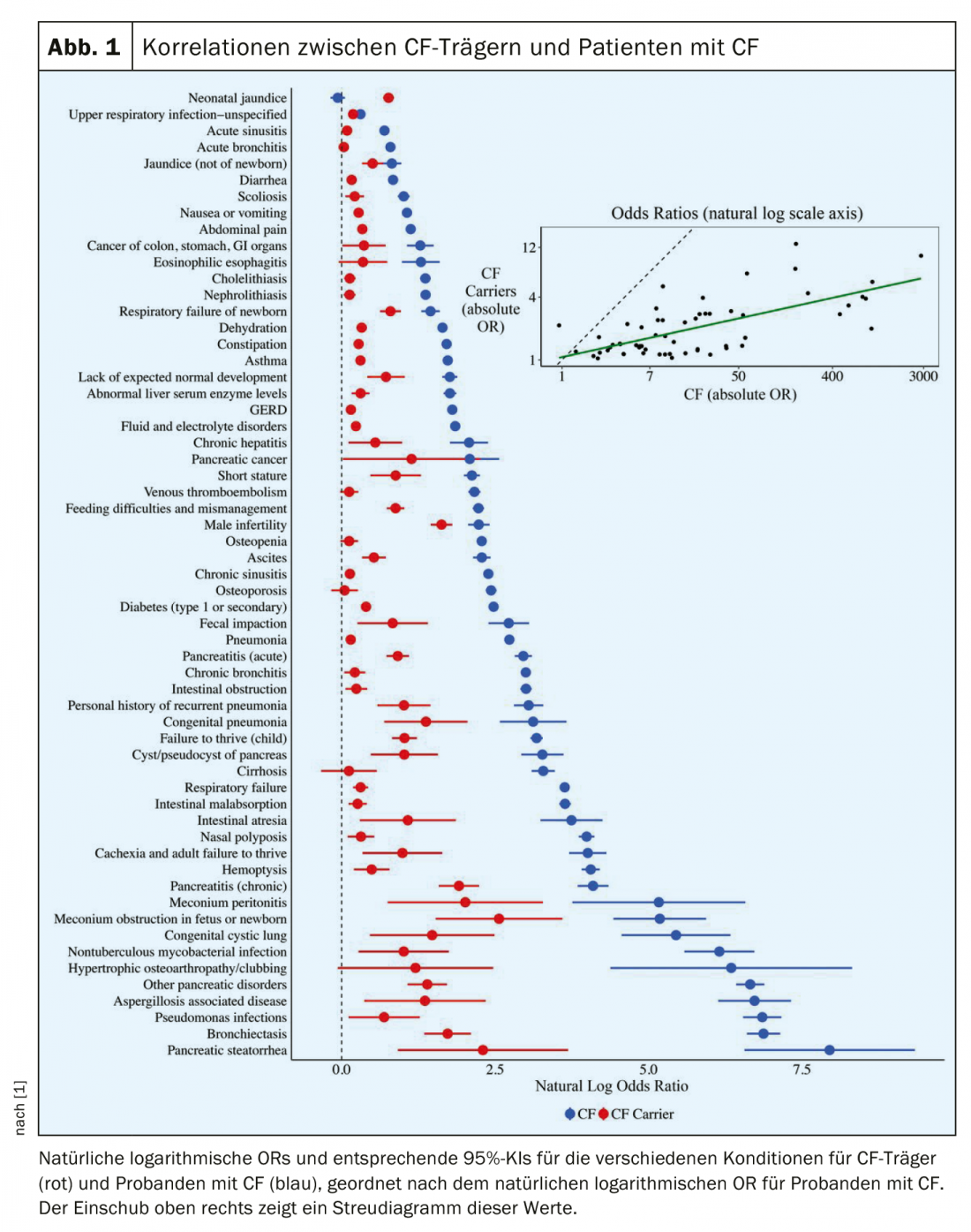
The aim of the study was to determine whether CF carriers are at increased risk for a range of CF-associated symptoms or diseases. The prevalence of diseases commonly associated with CF in large populations of CF carriers was examined. Also, the risk for each disease was compared between CF subjects and patients with CF. All 59 CF-associated diseases were more common in CF carriers compared with controls, with significantly increased risk for 57 (p<0.05). This concerned not only medical conditions whose association with CF carriers was previously known, such as. pancreatitis, male infertility, or bronchiectasis, but also conditions for which an association had not previously been reported, including type 1 diabetes (OR 1.49; 95% CI 1.40-1.59), constipation (OR 1.32; 95% CI 1.24-1.41), cholelithiasis (OR 1.14; 95% CI 1.04-1.25), childhood failure to thrive (OR 2.78; 95% CI 2.28-3.41), and short stature (OR 2.41; 95% CI 1.60-3.64). As the relative likelihood of a given condition in patients with CF increased, so did the corresponding relative likelihood for carriers (p<0.001).
Figure 1 shows the correlation in estimated natural log ORs across all conditions between CF subjects and patients with CF, ordered by ORs among patients with CF. In general, as the relative likelihood of a given condition increases for patients with CF, the corresponding relative probabilities for CF carriers also increase. The Pearson correlation coefficient between the natural log-ORs in the CF carrier and CF cohorts was 0.67 (p<0.001). Both CF carriers and patients with CF were significantly more likely than controls to have multiple CF-associated symptomatology affecting different organ systems. Also, in both CF carriers and patients with CF, ORs tended to increase with the number of CF-related diseases and organ systems affected.
The findings suggest that CF carriers are at increased risk of developing a variety of CF-related diseases across multiple organ systems, the authors write. This risk was observed in a cohort of CF carriers identified by genetic testing and in a secondary cohort of mothers of children with CF before the child was born with CF. CF carriers showed a similar, albeit attenuated, phenotype compared with individuals with CF. CF carriers were more likely than controls to suffer from multiple CF-related conditions. Taken together, Miller et al. said, the study’s findings challenge the assumption that CFTR expression levels associated with the CF carrier state are sufficient to fully protect subjects from CF-related conditions.
No cause for concern despite high ORs
Nevertheless, despite the relatively large ORs, the researchers see no reason for undue concern. To this end, they point out the difference between relative and absolute risk. For example, in a condition such as chronic pancreatitis, the relative risk for carriers is very high (OR 6.76; 95% CI 4.87-9.39), but the absolute risk even for CF carriers was low in their sample (0.429 per 100). For this reason, the vast majority of CF carriers will never develop pancreatitis.
More and more CF carriers are being identified. According to the study authors, knowing that carriers are at higher risk for CF-related diseases can help physicians and patients choose more effective screening and prevention approaches: For example, knowing that one is a CF carrier can motivate and help avoid other disease risk factors, such as excessive alcohol consumption, they say.
In addition, identifying patients with conditions such as pancreatitis who are also CF carriers may one day have implications for treatment, he said. Newer drugs developed to treat cystic fibrosis by correcting defects in CFTR function may increase Cl- and HCO3- transport. For example, the CFTR potentiator ivacaftor increases the likelihood of wild-type CFTR, and its effect on the nonmutant allele may be sufficient to attenuate or prevent the phenotype observed in CF carriers.
The catch to the study, as Miller et al. itself is limiting is the relatively small number of CF carriers that could be identified. And those carriers who were identified were predominantly young, limiting the ability to study risk for disease conditions that are more common in older populations (e.g., osteoporosis, cirrhosis). In addition, the data do not allow identification of specific CFTR mutations, and it is possible that the results may not be generalizable to all CF carriers. (However, most CF carriers have the F508del mutation). Further, the data available to the researchers also did not allow for direct analysis of demographic factors (e.g., race), and the results of the U.S. study may not be generalizable to other populations because all subjects had health insurance.
Nevertheless, the authors believe that identifying CF carriers may aid in the prevention, diagnosis, and treatment of several common and uncommon conditions. With increased genetic screening, she believes it may be possible to apply a similar approach to people who are carriers of other recessive genetic diseases.
Literature:
- Miller AC, et al: Proc Natl Acad Sci USA 2020; 117: 1621-1627.
InFo PNEUMOLOGY & ALLERGOLOGY 2020; 2(2): 18-20.