Paranodopathies are a new special form of inflammatory polyneuropathies, mediated by autoantibody-induced damage at Ranvier’s lacing ring. What are characteristics of this disease and how can it be treated?
Polyneuropathies are a heterogeneous group of disorders that result from damage to the axon or myelin sheath of peripheral nerves. Although the cause remains unclear in about 25% of all polyneuropathies, etiologic differentiation is essential in clinical practice [1,2]. In this way, treatable causes can be identified and specific therapies offered, especially in the immune neuropathies. Basic diagnostics in addition to history, examination and laboratory diagnostics include electroneurography, which can distinguish between an axonal and demyelinating pattern, and nerve ultrasound. In recent years, the new entity of paranodopathies has been described, which cannot be assigned to the classical concept of “axonal” and “demyelinating” [3–5]: Here, Ranvier’s lacing ring represents the starting point of the disease. Paranodal autoantibodies are valuable biomarkers in immune neuropathies because paranodopathies differ from other immune neuropathies in terms of response to therapy.
Inflammatory neuropathies
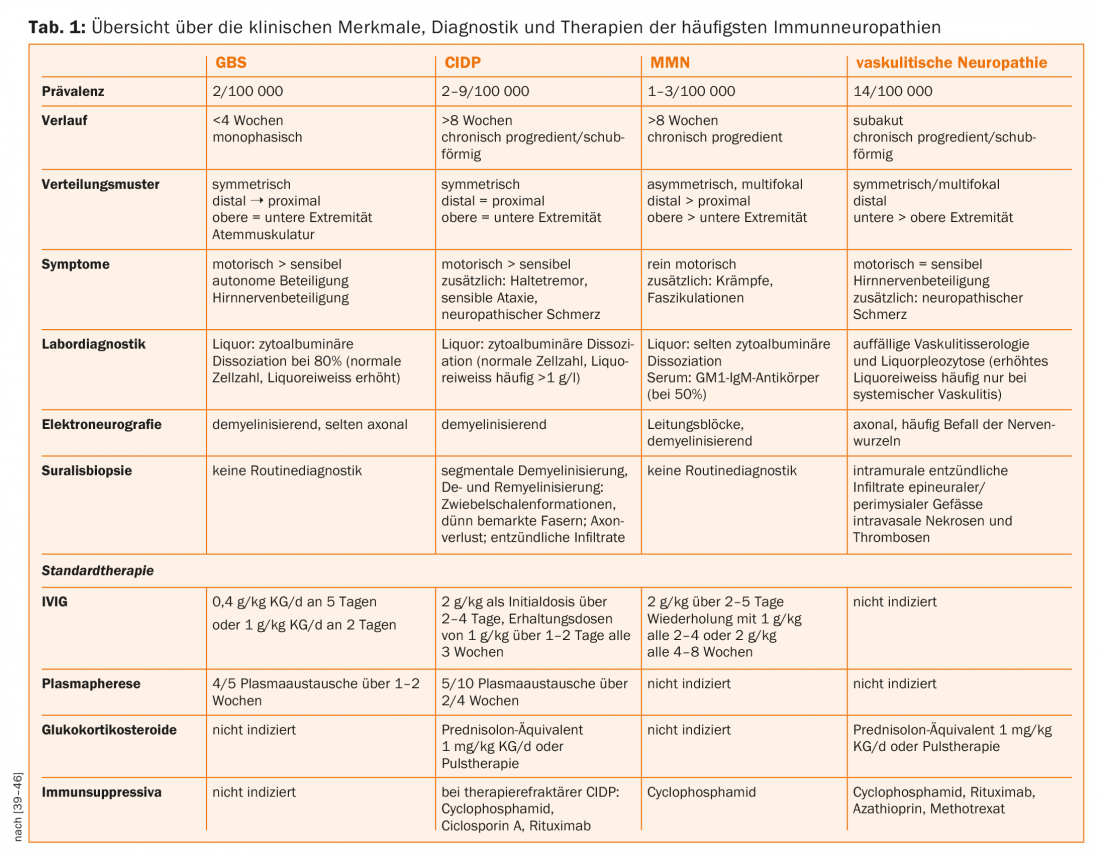
Inflammatory neuropathies include infectious neuropathies, which are rare in Central Europe, and autoimmune-mediated neuropathies, which, with a prevalence of about 10%, are the third most common cause of all polyneuropathies after diabetic and ethyltoxic polyneuropathies [2,6]. The prominent representatives of demyelinating forms are acute-onset Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), and multifocal motor neuropathy (MMN). In these diseases, the myelin sheath is damaged by humoral and cellular immune processes, and axonal loss may occur secondarily. In vasculitic neuropathy, on the other hand, ischemic axonal damage results from mostly isolated involvement of the vasa nervorum. For the differential diagnosis of immune neuropathies, the course, distribution pattern, laboratory diagnostics, and especially electrophysiology and nerve biopsy are crucial. The exact differentiation is particularly important with regard to the different therapy options (Tab. 1).
The paranodium as the starting point of rare immune neuropathies
The Ranvier’s cord ring of peripheral myelinated nerves is subdivided into three sections (Fig. 1). At the nodium, the voltage-gated sodium channels, which are responsible for saltatory excitation conduction, are bundled by neurofascin-186 [7]. The region is flanked by the paranodium, where the terminal myelin loops lie loosely around the axon. Here, the protein complex of glial neurofascin-155 and axonal contactin-1 and caspr-1 connects myelin and axon and is crucial for maintaining nodal architecture and normal nerve conduction velocities [8,9]. Lateral to this, voltage-gated potassium channels are concentrated at the juxtaparanodium by a protein complex of contactin-2 and caspr-2 [10].
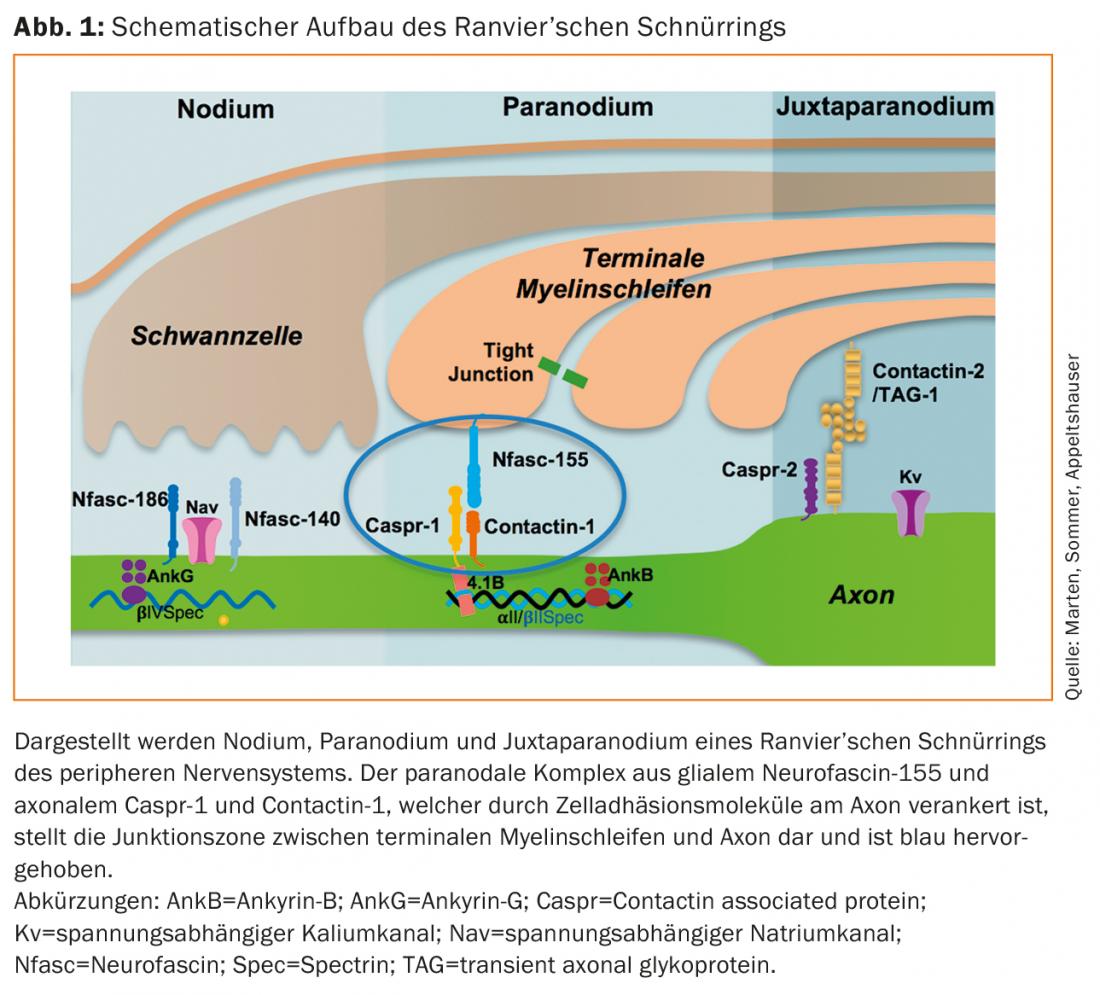
Several histopathological studies describe an alteration of the lacing ring architecture in various polyneuropathies [11–13]. In recent years, IgG autoantibodies and more recently IgM autoantibodies against the paranodal proteins neurofascin-155, contactin-1, and caspr-1 have been described in patients with immune neuropathies [14–20]. In most cases, these are autoantibodies of the complement-independent IgG subclass 4, but IgG3 autoantibodies that can bind and activate complement in vitro have also been described in patients with acute disease [16,21]. Electrophysiologically, these autoantibody-mediated immune neuropathies may meet criteria for CIDP or GBS, but not histopathologically. Here, there is no macrophage-dependent de- and remyelination, but (secondary) axonal damage and detachment of terminal myelin loops from the paranodal axon, as well as elongation of the nodes and dispersion of paranodal proteins and ion channels [14,16,22,23]. This architectural disruption of Ranvier’s lacing ring is thought to be responsible for conduction blocks and prolonged nerve conduction velocities in the paranodopathies [5,24]. Passive-transfer models confirm paranodal destruction and conduction blocks in vivo after injection of anti-contactin-1 IgG4 and anti-neurofascin-155 IgG autoantibodies [25,26]. Thus, the starting point of the disease is not the myelin sheath but the cord ring, which is why no assignment to the classical categories “axonal” or “demyelinating” is possible. Therefore, the term “paranodopathy,” which originally referred to anti-ganglioside autoantibody-associated immune neuropathies, has now been established to refer specifically to immune neuropathies with paranodal autoantibodies [3,4,22,27].
Clinical features, diagnostics and therapy
Paranodal autoantibodies have been identified almost exclusively in patients with the clinical presentation of CIDP and GBS. Here, the prevalences are mostly <10% [28–30]. Affected patients show acute or subacute onset of disease and develop severe motor involvement. In addition, a pronounced tremor and sensory ataxia can aggravate the clinical course [31,32]. IgG4 antibodies to neurofascin-155 have the highest prevalence [33]. In these seropositive patients, central demyelination foci, tremor, and cerebellar ataxia have also been described as possible signs of central autoantibody-mediated damage. The pathomechanism and prevalence of anti-neurofascin-155 IgG autoantibodies in patients with clinically combined central and peripheral involvement have not been adequately studied and are controversial in the literature [30,33–36]. Autoantibodies to Caspr-1 have rarely been detected, with IgG4 autoantibodies described in patients with a chronic course and IgG3 autoantibodies in a patient with GBS [16,37]. Neuropathic pain has been proposed as a common characteristic with histopathological correlate, namely binding of autoantibodies to nociceptive spinal ganglion neurons. However, larger clinical and experimental studies to confirm this are lacking [16]. Table 2 gives an overview of the autoantibody-specific characteristics of the paranodopathies.
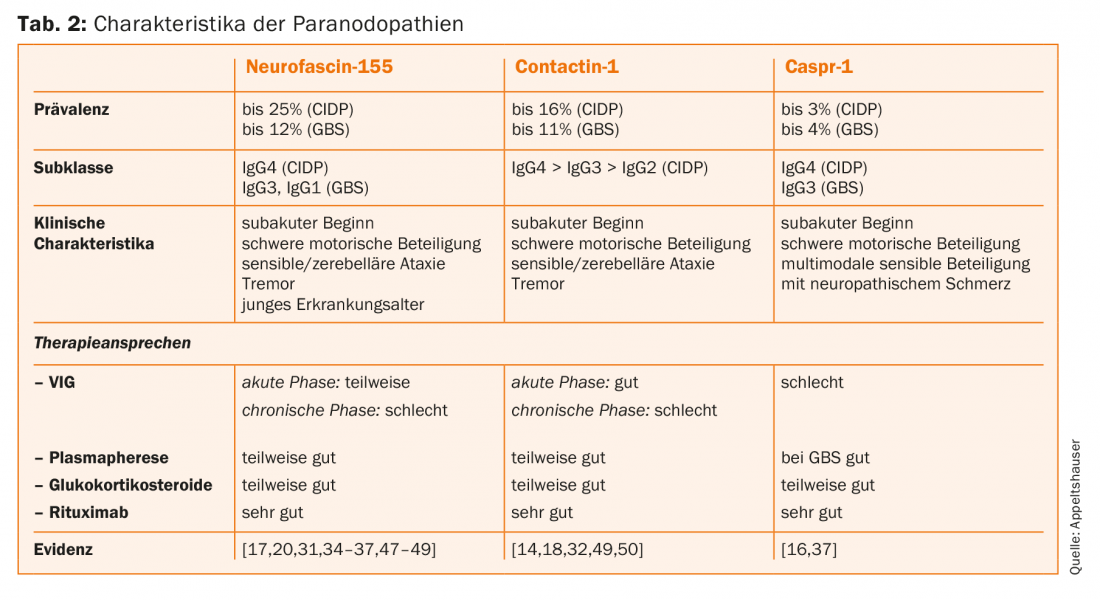
The autoantibodies can be detected in serum by ELISA as well as binding assays on mouse or rat pluck nerves and transfected cells (Fig. 2) [33]. No validated commercial test is available yet, but testing is possible in specialized research laboratories. Screening is only useful in patients with the typical clinical phenotype. In the case of mild, chronic, distal sensory-motor neuropathies, on the other hand, clarification of the common causes is useful.
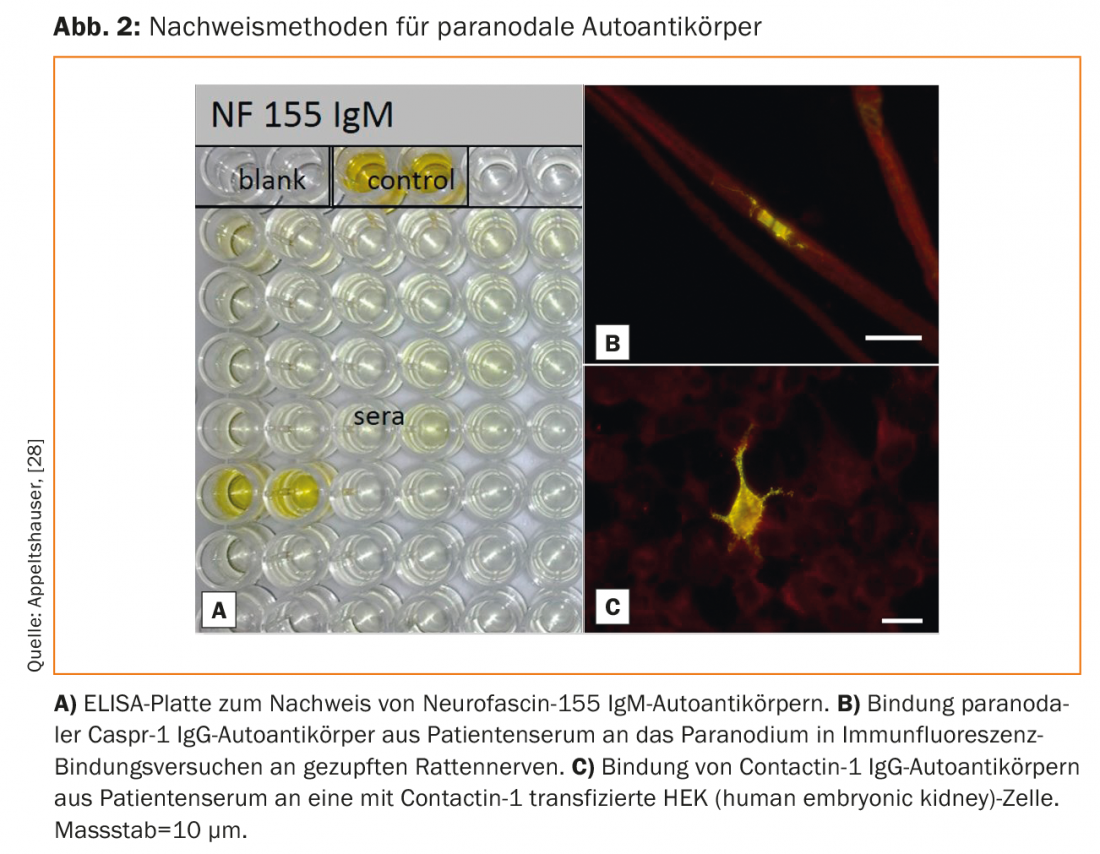
Because of the low number of cases of patients with paranodopathies, there are no controlled therapy studies so far. Case series indicate that, in contrast to classical CIDP, patients with IgG4-mediated paranodopathy respond poorly, whereas patients with IgG3-mediated paranodopathy respond quite well to IVIG [14,21,30].
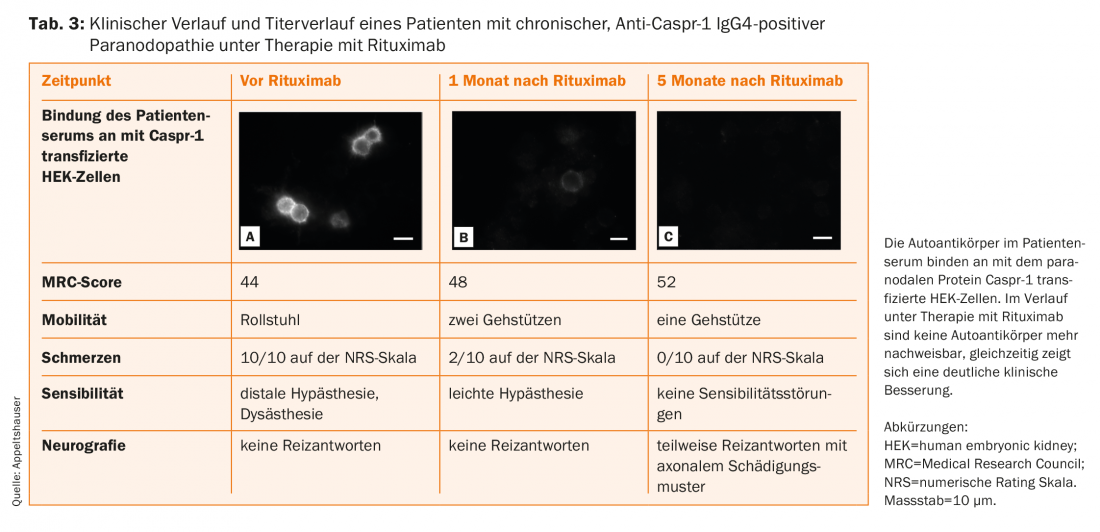
Patients with IgG4 autoantibodies respond very well to rituximab in the case series, with a significant reduction in antibody titers found as a surrogate parameter (Table 3) [14,16,20,38]. It is possible that early initiation of therapy may prevent chronic axonal injury. Therefore, the current guideline also recommends an individual therapy trial with rituximab in case of detection of paranodal IgG4 autoantibodies, which should be performed under close follow-up at specialized centers [39]. Furthermore, follow-up of patients with paranodopathies is essential for a better understanding of the disease of this new entity.
Take-Home Messages
- The etiology of inflammatory polyneuropathies is heterogeneous and differs between axonal and demyelinating polyneuropathies.
- The paranodopathies are a new special form of inflammatory polyneuropathies, mediated by autoantibody-induced damage at Ranvier’s lacing ring.
- Clinical features of paranodopathies include: subacute onset, severe motor involvement, poor response to standard therapies, tremor, and young age of onset for autoantibodies to neurofascin-155, plus ataxia for autoantibodies to contactin-1 and pain for antibodies to caspr-1.
- Identification of patients with paranodal autoantibodies is important for a better understanding of the disease and for treatment decisions.
Literature:
- Pasnoor M, et al: Cryptogenic sensory polyneuropathy. Neurol Clin 2013; 31(2): 463-476.
- Visser NA, et al: Incidence of polyneuropathy in Utrecht, the Netherlands. Neurology 2015; 84(3): 259-264.
- Uncini A: A common mechanism and a new categorization for anti-ganglioside antibody-mediated neuropathies. Exp Neurol 2012; 235(2): 513-516.
- Uncini A, Vallat JM: Autoimmune nodo-paranodopathies of peripheral nerve: the concept is gaining ground. J Neurol Neurosurg Psychiatry 2017; 89(6): 627-635.
- Uncini A, Susuki K, Yuki N: Nodo-paranodopathy: beyond the demyelinating and axonal classification in anti-ganglioside antibody-mediated neuropathies. Clin Neurophysiol 2013; 124(10): 1928-1934.
- Hanewinckel R, et al: Prevalence of polyneuropathy in the general middle-aged and elderly population. Neurology 2016; 87(18): 1892-1898.
- Stathopoulos P, Alexopoulos H, Dalakas MC: Autoimmune antigenic targets at the node of Ranvier in demyelinating disorders. Nat Rev Neurol 2015; 11(3): 143-156.
- Vallat JM, et al: Contactin-Associated Protein 1 (CNTNAP1) Mutations Induce Characteristic Lesions of the Paranodal Region. J Neuropathol Exp Neurol 2016; 75(12): 1155-1159.
- Sherman DL, et al: Neurofascins are required to establish axonal domains for saltatory conduction. Neuron 2005; 48(5): 737-742.
- Poliak S, et al: Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 1999; 24(4): 1037-1047.
- Doppler K, et al: Disruption of nodal architecture in skin biopsies of patients with demyelinating neuropathies. J Peripheral Nerv Syst 2013; 18(2): 168-176.
- Li J, et al: Skin biopsies in myelin-related neuropathies: bringing molecular pathology to the bedside. Brain 2005; 128(5): 1168-1177.
- Cifuentes-Diaz C, et al: Nodes of ranvier and paranodes in chronic acquired neuropathies. PloS one 2011; 6(1): e14533.
- Doppler K, et al: Destruction of paranodal architecture in inflammatory neuropathy with anti-contactin-1 autoantibodies. J Neurol Neurosurg Psychiatry 2015; 86(7): 720-728.
- Devaux JJ, et al: Antibodies to gliomedin cause peripheral demyelinating neuropathy and the dismantling of the nodes of Ranvier. Am J Pathol 2012; 181(4): 1402-1413.
- Doppler K, et al: Auto-antibodies to contactin-associated protein 1 (Caspr) in two patients with painful inflammatory neuropathy. Brain 2016; 139(10): 2617-2630.
- Ng JK, et al: Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology 2012; 79(23): 2241-2248.
- Devaux JJ, Odaka M, Yuki N: Nodal proteins are target antigens in Guillain-Barre syndrome. J Peripheral Nerve Syst 2012; 17(1): 62-71.
- Doppler K, et al: Neurofascin-155 IgM autoantibodies in patients with inflammatory neuropathies. J Neurol Neurosurg Psychiatry 2018; 89(11): 1145-1151.
- Burnor E, et al: Neurofascin antibodies in autoimmune, genetic, and idiopathic neuropathies. Neurology 2018; 90(1): e31-e38.
- Appeltshauser L, et al: Complement deposition induced by binding of anti-contactin-1 auto-antibodies is modified by immunoglobulins. Exp Neurol 2017; 287(1): 84-90.
- Koike H, et al: Paranodal dissection in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin-155 and anti-contactin-1 antibodies. J Neurol Neurosurg Psychiatry 2017; 88(6): 465-473.
- Vallat JM, et al: Subacute nodopathy with conduction blocks and anti-neurofascin 140/186 antibodies: an ultrastructural study. Brain 2018; 141(7): e56.
- Vallat JM, et al: Paranodal lesions in chronic inflammatory demyelinating polyneuropathy associated with anti-neurofascin 155 antibodies. Neuromuscul Disord 2017; 27(3): 290-293.
- Manso C, et al: Contactin-1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain 2016; 139(6): 1700-1712.
- Yan W, et al: Antibodies to neurofascin exacerbate adoptive transfer experimental autoimmune neuritis. J Neuroimmunol 2014; 277(1-2): 13-17.
- Kuwabara S, Misawa S, Mori M: Nodopathy: chronic inflammatory demyelinating polyneuropathy with anti-neurofascin 155 antibodies. J Neurol Neurosurg Psychiatry 2017; 88(6): 459.
- Doppler K, et al: Contactin-1 and Neurofascin-155/-186 Are Not Targets of Auto-Antibodies in Multifocal Motor Neuropathy. PloS one 2015; 10(7): e0134274.
- Doppler K, Sommer C: New entity of paranodopathies: a target structure with therapeutic consequences. Act Neurol 2017; 44(3): 194-199.
- Hu W, et al: Association of neurofascin IgG4 and atypical chronic inflammatory demyelinating polyneuropathy: A systematic review and meta-analysis. Brain Behav 2018: 8(10): e1115.
- Querol L, et al: Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology 2014; 82(10): 879-886.
- Miura Y, et al: Contactin 1 IgG4 associates to chronic inflammatory demyelinating polyneuropathy with sensory ataxia. Brain 2015; 138(6): 1484-1491.
- Vural A, Doppler K, Meinl E: Autoantibodies Against the Node of Ranvier in Seropositive Chronic Inflammatory Demyelinating Polyneuropathy: Diagnostic, Pathogenic, and Therapeutic Relevance. Front Immunol 2018; 9: 1029.
- Kawamura N, et al: Anti-neurofascin antibody in patients with combined central and peripheral demyelination. Neurology 2013; 81(8): 714-722.
- Devaux JJ, et al: Neurofascin-155 IgG4 in chronic inflammatory demyelinating polyneuropathy. Neurology 2016; 86(9): 800-807.
- Ogata H, et al: Characterization of IgG4 anti-neurofascin 155 antibody-positive polyneuropathy. Ann Clin Transl Neurol 2015; 2(10): 960-971.
- Delmont E, et al: Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain 2017; 140(7): 1851-1858.
- Querol L, et al: Rituximab in treatment-resistant CIDP with antibodies against paranodal proteins. Neurol Neuroimmunol Neuroinflamm 2015; 2(5): e149.
- Sommer C, et al: Therapy of acute and chronic immune-mediated neuropathies and neuritides, S2e guideline, 2018. In: Commission Guidelines of the German Society of Neurology: Guidelines for Diagnostics and Therapy in Neurology.
- Vedeler CA, Farbu E, Mellgren SI: Chronic inflammatory demyelinating polyneuropathy (CIDP). Acta Neurol Scand Suppl 2013; 196: 48-51.
- Saperstein DS, et al: Clinical spectrum of chronic acquired demyelinating polyneuropathies. Muscle Nerve 2001; 24(3): 311-324.
- Muley SA, Parry GJ: Multifocal motor neuropathy. J Clin Neurosci 2012; 19(9): 1201-1209.
- Sommer C, et al: Polyneuropathies. Dtsch Arztebl Int 2018; 115(6): 83-90.
- Hadden RDM, et al: Vasculitic peripheral neuropathy: Case definition and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine 2017; 35(11): 1567-1578.
- Collins MP, Hadden RD: The nonsystemic vasculitic neuropathies. Nat Rev Neurol 2017; 13(5): 302-316.
- Callaghan BC, et al: The Importance of Rare Subtypes in Diagnosis and Treatment of Peripheral Neuropathy: A Review. JAMA Neurol 2015; 72(12): 1510-1518.
- Kadoya M, et al: IgG4 anti-neurofascin155 antibodies in chronic inflammatory demyelinating polyradiculoneuropathy: Clinical significance and diagnostic utility of a conventional assay. J Neuroimmunol 2016; 301: 16-22.
- Cortese A, et al: Neurofascin-155 as a putative antigen in combined central and peripheral demyelination. Neurol Neuroimmunol Neuroinflamm 2016; 3(4): e238.
- Mathey EK, et al: Autoantibody responses to nodal and paranodal antigens in chronic inflammatory neuropathies. J Neuroimmunol 2017; 309: 41-46.
- Querol L, et al: Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 2013; 73(3): 370-380.
InFo NEUROLOGY & PSYCHIATRY 2018; 16(6): 14-18.