Amyloidoses are a rare form of deposit dermatoses caused by defective folding of proteins. Dermatological early detection is of great importance for the further course of the disease.
Etymologically, the term amyloidoses goes back to ancient Greek ἄμυλον ámylon, meaning “power meal, starch.” Since biochemically a protein structure is present, this is a misleading name [1]. Amyloids are a group of heterogeneous proteins that appear ultrastructurally similar or react similarly on histological staining. Both localized and systemic amyloidoses may be characterized by cutaneous symptoms. “More than 25 proteins are capable of forming amyloid in healthy humans,” said Antonio Cozzio, MD, Clinic for Dermatology, Venereology and Allergology at the Kantonsspital St.Gallen, who spoke on this topic at this year’s Zurich Dermatology Training Days. Of the biochemically diverse proteins capable of forming amyloid, 15 can lead to clinically relevant disease [2].

Taxonomy in transition
Early detection by the dermatologist provides an important basis for effective treatment and prevention of further deterioration of any organs involved [3]. In 2016, the classification system of the Society for Amyloidoses was revised [4]: Congophilia and birefringence are still the main classificatory criteria. In addition, protein sequence analysis is recommended to determine the chemical identity of amyloid fibril protein deposited in the extracellular space of tissues. To date, 36 extracellular fibrillar pro- teins are known in humans, 2 of these are iatrogenic in nature, and 9 have also been identified in animals. Two recently discovered fibrillar proteins are AApoCII (derivative of apolipoprotein CII) and AApoCIII (derivative of apolipoprotein CIII). AApoCII and AApoCIII amyloidosis are hereditary systemic amyloidoses. Two proteins previously considered intracellular inclusions, tau and α-synuclein, are now taxed as extracellular deposits associated with cell death, designated “ATau” and “AαSyn”.
Heterogeneous cutaneous involvement
A distinction is made between systemic and cutaneous limited forms of amyloidosis, each of which is subdivided into acquired and hereditary subtypes (Fig. 1).
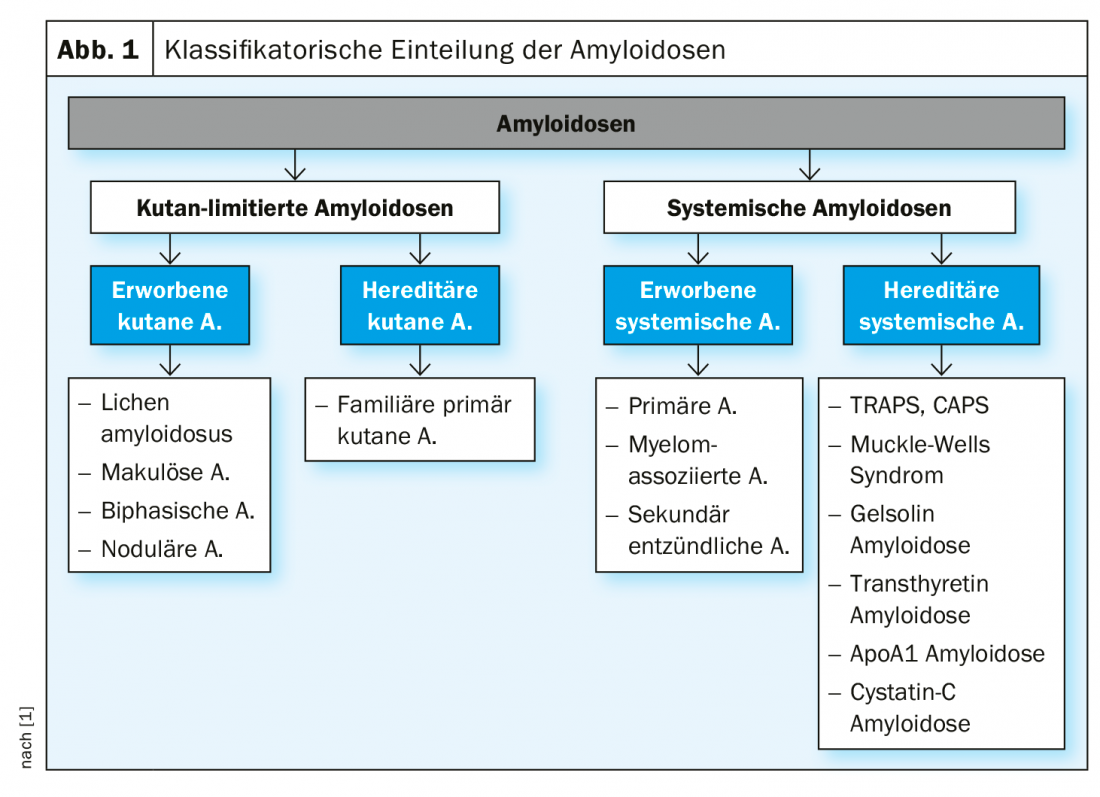
Systemic amyloidosis: This is a heterogeneous clinical picture characterized by the deposition of endogenous, pathologically altered proteins in tissue or organs. Cutaneous signs characteristic of various forms of acquired systemic amyloidosis are summarized in Table 1.
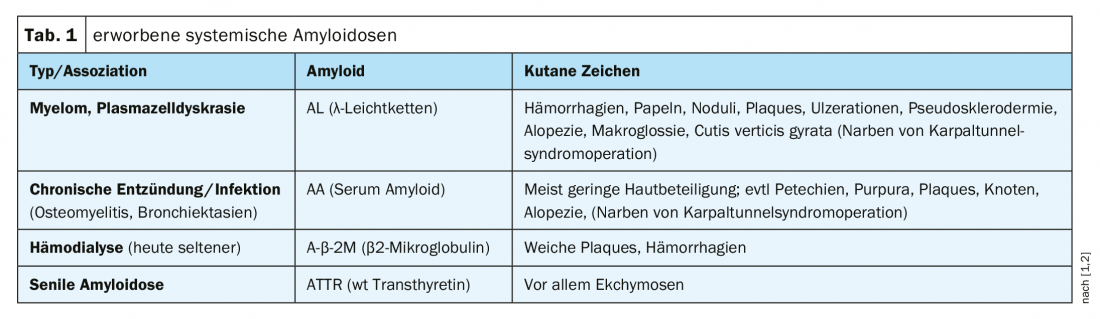
Light chain amyloidosis is the most common form and is often very aggressive. If the heart or kidneys are affected, this form of amyloidosis can lead to death after a few months if left untreated. Antibody light chains are thought to play an important role in the etiology, leading to the characteristic tissue deposits. Systemic amyloidosis is common in patients with multiple myeloma. In an early stage of primary systemic amyloidosis, clinical manifestations are initially limited to dermatologic symptoms, with organ involvement occurring later [5]. The diagnostic spectrum includes tissue and bone marrow biopsy, electrophoresis, and organ-specific examinations (Overview 1).


Cutaneous limited amyloidosis: This collective term refers to dermatologic pathologies that are histologically characterized by extracellular accumulation of amyloid deposits in the dermis. “Macular and papular amyloidoses are usually cutaneously limited,” the speaker explained. If nodal cutaneous amyloidosis is suspected, it should be clarified whether cardiac/renal involvement is present; collaboration with hematooncology may be required [1]. In case of unclear purpura (especially in case of additional nodularity, macroglossia), a biopsy with immunohistochemical analysis is recommended, and histological findings in the history may also be included. The following differential diagnoses should be excluded: cutaneous neoplasms, sarcoidosis, granuloma faciale, leishmaniasis (review 2) [1]. The range of treatment options is relatively broad and should be individualized (box).
Literature:
- Cozzio A: Slide presentation: annual theme of depositional dermatoses. Amyloidoses, Antonio Cozzio, MD, 9th Zurich Dermatology Training Days 2019, Zurich, June 26, 2019.
- Rauch PJ: Systemic amyloidoses. Switzerland Med Forum 2014; 14(50): 943-948.
- Bruch-Gerharz D, Ruzicka T: Amyloidoses and hyalinoses. In: Plewig G., Ruzicka T., Kaufmann R, Hertl M (eds) Braun-Falco’s Dermatology, Venereology and Allergology 2017. Springer Reference Medicine. Berlin, Heidelberg: Springer.
- Sipe JD, et al: Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. The Journal of Protein Folding Disorders
- Trajber Horvat A, Trčko K, Jurčić V, Marko PB: Primary Systemic Amyloidosis With Skin and Cardiac Involvement: A Case Report. Acta Dermatovenerologica Alpina, Pannonica, et Adriatica 2018. www.oneamyloidosisvoice.com/rcuratenew
DERMATOLOGIE PRAXIS 2019; 29(5): 27-28 (published 10/10/19, ahead of print).